




























Step 6 of 7
Résistance aux antiplaquettaires
Tous les patients ne réagissent pas avec la même intensité aux antiplaquettaires. Les faibles répondeurs sont particulièrement fréquents avec l’aspirine (3-20% des patients) et avec le clopidogrel (12-30%). Ce taux varie sur une vaste fourchette parce qu’il dépend de plusieurs phénomènes [12,17,22].
- Type de test de fonction plaquettaire utilisé (voir Tests d’activité plaquettaire) ; chaque test mesure une activité différente des plaquettes ; la sensibilité et la spécificité varie selon le récepteur concerné, ce qui rend la comparaison des tests très difficile. Il n’existe pas de test standardisé, mais des valeurs-seuils de référence ont été adoptées très récemment [3].
- Polymorphisme génétique (voir Génomique) : des modifications génétiques dans le codage des enzymes liés à l’absorption et au métabolisme du clopidogrel en altèrent l’efficacité [45].
- Interactions médicamenteuses (voir ci-dessous) : plusieurs substances interfèrent avec le métabolisme du clopidogrel et diminuent la production de métabolite actif [4].
- Variations enzymatiques ; le diabète type I s’accompagne d’une hyperactivité des estérases plasmatiques et tissulaires qui transforment le clopidogrel en un métabolite inactif [1]. Au contraire, la fumée de cigarette et la cafféine sont des inducteurs du cytochrome P450 ; elles accélèrent la biotransformation du clopidogrel en son métabolite actif [34].
- Variations au niveau cellulaire : turnover accéléré des plaquettes, surexpression des récepteurs P2Y1 et P2Y12, excès de noradrénaline, métabolisme extraplaquettaire de l’aspirine [16].
- Variation spontanée de la réactivité plaquettaire dans le temps ; certains individus modifient leur réponse aux antiplaquettaires d’une semaine à l’autre [37].
- Polymorbidité : certaines affections s’accompagnent d’une hyperactivité plaquettaire qui diminue l’efficacité des médicaments : diabète (turnover plaquettaire accéléré), SIRS, stress opératoire, hypercholestérolémie, insuffisance rénale, syndrome paranéoplasique, âge avancé [22].
- Non-compliance du patient ; jusqu’à 22% de la population ne prend pas régulièrement ses médicaments, ou adapte la prescription de manière inappropriée [38].
Aspirine
Le taux de faibles répondeurs à l’aspirine est particulièrement important chez les diabétiques de type I et dans le sexe féminin [24]. L’incidence varie de 3% à 27% en fonction du test utilisé. Avec un test spécifique pour la COX-1 (test avec l’acide arachidonique comme agoniste), l’incidence n’est que de 3% ; avec un test non-spécifique, elle est de 12-15% [31]. Le taux moyen de non-répondeurs se situe probablement aux environs de 6% de la population [8]. Les individus dits résistants à l’aspirine ont souvent un blocage efficace de la production de TXA2, mais une prédominance des autres voies de stimulation du récepteur GP-IIb/IIIa (récepteur ADP, récepteur à la thrombine, etc) ; ils conservent ainsi une agrégabilité plaquettaire importante malgré l’activité inhibitrice de l’aspirine. Une "résistance" à l’aspirine ne justifie pas une augmentation du dosage (75-160 mg) [3].
Clopidogrel
La réponse au clopidogrel suit une répartition gaussienne ; la majorité des patients présente un degré d’inhibition plaquettaire de 40-60%, avec de faibles répondeurs d’un côté et quelques hyper-répondeurs de l’autre (Figure 29.6) [39].

Figure 29.6 : Répartition du degré d’inhibition plaquettaire chez 544 patients recevant 75 mg/j de clopidogrel [39]. La majorité des patients présente un degré d’inhibition plaquettaire de 40-60%. Les forts répondeurs ont un risque hémorragique plus élevé que la moyenne (HR 2.6) [42].
La moitié des malades qui ne sont pas normalement sensibles à l’aspirine ne répond pas non plus au clopidogrel [15]. La pharmacologie de ce dernier est surtout caractérisée par des interactions avec d’autres médicaments et par un très important polymorphisme génétique.
Interactions médicamenteuses
Les anti-inflammatoires non-stéroïdiens bloquent l’accès de l’aspirine à la COX-1 qu’ils inhibent, mais n’ont pas d’activité antiplaquettaire [14] ; ainsi l’association d’ibuprofen diminue de 70% la protection cardiovasculaire de l’aspirine (HR 1.73) [25].
De nombreuses substances entrent en compétition avec le clopidogrel pour leur métabolisme par les cytochromes hépatiques du groupe P450 : statines lipophiles, certains inhibiteurs de la pompe à proton (IPP), midazolam, bloqueurs calciques, fluoxétine, kétoconazole, cimétidine. Cette compétition peut diminuer l’efficacité clinique du clopidogrel de 25% [2,4,13,18,33]. Deux substances sont particulièrement concernées par cette interférence.
- L’atorvastatine, une statine liposoluble, augmente le risque cardiovasculaire chez les patients sous clopidogrel [4] ; bien que cet impact n’apparaissent que dans certaines études, sa probabilité est considérée comme significative par la FDA.
- L’oméprazole est lié à une augmentation des complications cardio-vasculaires dans plusieurs études rétrospectives [13,18], mais non dans les méta-analyses ni dans les études randomisées [5,9,32,44] ; par contre, il diminue le taux d’hémorragies digestives [5,32].
Ces effets négatifs sur le devenir des patients n’ont jamais été retrouvés ni avec les statines hydrosolubles ni avec les autres IPP [23,44]. Ils ne concernent probablement que les malades qui sont à haut risque de thrombose ou qui n’ont qu’un faible degré d’inhibition plaquettaire. Malgré ces discordances, il est recommandé d’éviter la prescription d’atorvastatine et d’omeprazole chez les patients sous clopidogrel. Les autres IPP sont une aide précieuse pour diminuer le risque d’hémorragie digestive sous bi-thérapie [10,35].
Génomique
La pharmacogénomique démontre que les variations de l’activité plaquettaire en réponse au clopidogrel sont liées, entre autres, à des modifications dans le codage génétique situées à trois niveaux différents [26,45] :
- Absorption digestive (gène ABCB1) ;
- Métabolisme hépatique (gènes CYP3A5 et CYP2C19) ;
- Fonction des récepteurs plaquettaires P2Y12 et GP-IIb/IIIa (gène ITGB3).
Pour chacun de ces gènes, il existe des variantes (allèles) donnant naissance à des enzymes ou à des récepteurs non-fonctionnels. Pour les porteurs de ces allèles, l’efficacité du clopidogrel est diminuée, chez les homozygotes davantage que chez les hétérozygotes [26]. Les premières études avaient montré que la mortalité de ces porteurs et leur taux d’accidents cardiovasculaires 1 an après un événement coronarien étaient majorés par rapport aux malades dont les gènes sont normaux [27,30,45] :
- Allèles ABCB1 : HR 1.72 ;
- Allèles CYP2C19 : HR 1.98 – 4.04 ;
- Allèles ITGB3 : HR 3,69.
On a retrouvé le même phénomène avec l’aspirine ; les malades hétérozygotes sur certains récepteurs (Pl41/42) conservent une agrégabilité plaquettaire marquée sous des doses d'aspirine qui bloquent l'agrégation chez les porteurs de gènes normaux [8]. Les porteurs des variantes CYP2C19*2 et CYP2C19*3 du cytochrome hépatique, qui représentent 15% de la population blanche, 17% des afro-américains et 30% des asiatiques [11], ont un risque doublé de décès et de récidive d’infarctus ou d’ictus (OR 1.53-2.4), parce que le clopidogrel est insuffisamment transformé en son métabolite actif ; leur risque de thrombose de stent est 3 à 6 fois plus élevé (HR 3.09 – 6.02) (Figure 29.7) [27,40,45].
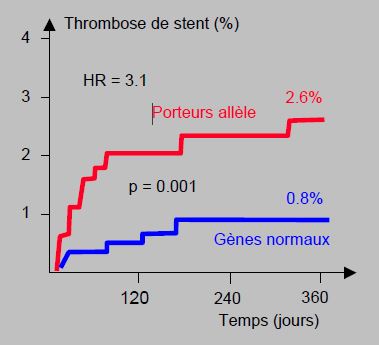
Figure 29.7 : Association entre la présence d’un allèle non-fonctionnel du gène CYP2C19 et la thrombose de stent chez 1'459 patients sous clopidogrel [27]. Le risque est trois fois plus élevé chez les malades dont le métabolisme hépatique du clopidogrel est déficient. HR : hazard ratio.
Chez les hétérozygotes pour l’allèle CYP2C19*2, l’inhibition plaquettaire peut être rétablie au niveau de celle des individus normaux en triplant la dose quotidienne de clopidogrel, mais cette astuce ne fonctionne pas chez les homozygotes [29]. Ces derniers peuvent bénéficier d’un traitement de prasugrel [28], car celui-ci permet de vaincre la non-réactivité plaquettaire chez la plupart des malades porteurs d’allèles hypométabolisateurs du clopidogrel [36]. Une méta-analyse des études sur l’impact clinique du polymorphisme génétique indique que les allèles sont probablement associés aux complications cardiovasculaires chez les malades à haut risque : après SCA, la mortalité et l’infarctus sont accrus de 75% (HR 1.76), la thrombose de stent est 4 fois plus élevée (HR 3.97) [30]. A telle enseigne que la FDA a un temps recommandé le génotypage CYP2C19 chez les malades à haut risque de thrombose de stent, puisque ce test est disponible sur le marché (coût : 500 $) [19,36].
Toutefois, d’autres rapports n’ont pas confirmé cette association : le mécanisme du CYP2C19*2 n’explique que le 12% de la variabilité de réponse au clopidogrel [39,46] et a une valeur prédictive positive de seulement 12-20% [27]. D’autre part, il n’a aucune valeur prédictive sur le risque hémorragique [19]. La question de savoir si le génotypage a un impact significatif sur le devenir des patients, en-dehors de ceux qui sont à haut risque de thrombose de stent, reste largement ouverte [20,21]. On ne saurait donc recommander la généralisation de ce test, d’autant plus que le polymorphisme génétique n’a pas d’impact sur le devenir des patients traités avec du ticagrelor ou du prasugrel [28,43].
Le risque hémorragique est directement lié à l’efficacité antiplaquettaire ; à taux équivalents de métabolite actif et d’inhibition plaquettaire, les saignements sont identiques avec les différentes thiénopyridines [6]. Les patients répondeurs sont mieux protégés contre les accidents cardiaques, mais sont aussi ceux qui ont le plus d’épisodes de pertes sanguines. Il existe également des hyper-répondeurs, porteurs de l’allèle CYP2C19*17, qui ont trois fois plus de saignements que les normo-répondeurs (OR 3.41), bien qu’ils présentent le même risque de thrombose de stent [41]. Toutefois, les tests génétiques échouent à déterminer quels sont les patients à risque accru de saignement [19]. Cette linéarité entre l'effet thérapeutique et le risque hémorragique des thiénopyridines, bloqueurs irréversibles du récepteur P2Y12, ne se retrouve pas au même degré avec les bloqueurs réversibles de ce même récepteur, comme le ticagrelor ou le cangrelor, où les deux effets sont partiellement dissociés.
Les tests génétiques prennent davantage de valeur prédictive lorsqu’on les combine à des éléments cliniques du pronostic. Par exemple, l’association des allèles hyporépondeurs de 3 gènes (ABCB1, CYP2C19 et ITGB3) avec la dose de charge en clopidogrel, la prescription d’IPP, le diabète, la fonction ventriculaire et les données de l’angioplastie (caractère aigu ou non, complexité des lésions coronaires) donne un ensemble qui prédit un risque de thrombose de stent précoce plus de 7 fois supérieur à la moyenne (HR 7.63) chez les malades avec des scores élevés dans chaque catégorie (Figure 29.8) [7].
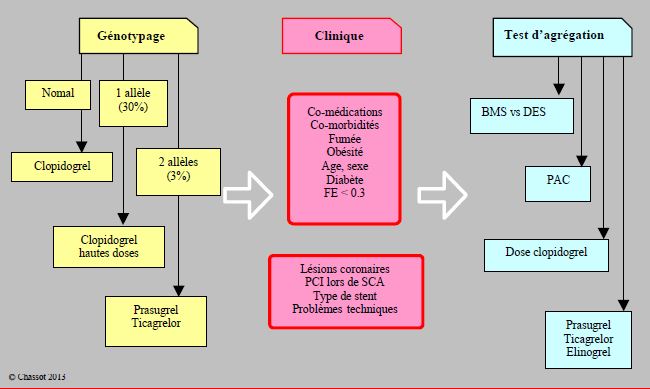
Figure 29.8 : Illustration schématique de l’impact possible du génotypage et des tests de fonction plaquettaire combinés au status clinique [d’après réf 7]. Le génotypage peut se pratiquer en-dehors de tout événement ischémique aigu. Il analyse les constituants stables et permanents de l’organisme, mais il ne représente que < 20% de la variabilité dans la réponse aux antiplaquettaires. Les sujets hétérozygotes pour l’allèle hyporépondeur (CYP2C19*2) peuvent bénéficier d’une dose élevée de clopidogrel (la tendance actuelle est plutôt d’abandonner cette option), mais non les homozygotes ; ces derniers ont avantage à recevoir du prasugrel ou du ticagrelor. Le génotypage représente l’arrière-fond sur lequel se surimpose la situation clinique, comme les comorbidités, les co-médications et les évènements coronariens. Les tests d’agrégation plaquettaire mesurent la réponse à une dose de charge de la bithérapie, et de ce fait évaluent la réponse réelle du patient dans son contexte clinique. Ils offrent des critères de choix pour le type de stent (les BMS sont préférables aux DES de 1ère génération chez les hyporépondeurs), pour l’indication aux PAC (préférence chez les faibles répondeurs), ou pour opter en faveur d’autres agents (prasugrel, ticagrelor, bientôt elinogrel). Comme on procède à ces tests après la dose de charge, le changement de stratégie présente évidemment certaines difficultés.
Malgré les espoirs qu’ils ont suscités, les tests génomiques ne se sont pas répandus dans la pratique clinique, d’autant plus que l’efficacité du ticagrelor, qui s’est imposé dans le traitement du SCA et la pose de stent, n’est pas influencée par le polymorphisme génétique.
| Résistance aux antiplaquettaires |
|
Le taux de non-réponse est d’environ 6% pour l’aspirine et 12-30% pour le clopidogrel. Plusieurs phénomènes interviennent :
- Interactions médicamenteuses (atorvastatine et oméprazole avec le clopidogrel) ;
- Variations enzymatiques (diabète) ;
- Hyperactivité plaquettaire (diabète, insuffisance rénale, SIRS, âge avancé) ;
- Non-compliance des patients (22% de la population) ;
- Polymorphisme génétique ;
- Test utilisé pour l’évaluation.
Les porteurs d’allèles codant des enzymes ou des récepteurs inactifs dans l’absorption intestinale, le métabolisme hépatique et l’action du clopidogrel sur les récepteurs plaquettaires présentent un risque de thrombose de stent plus élevé après SCA que les porteurs de gènes normaux. Cependant, la génomique n’explique que 12-20% de la variabilité de réponse au clopidogrel. D’autre part, l’activité du prasugrel et du ticagrelor est indépendante du polymorphisme génétique. L’utilisation massive de ces deux substances dans les cas à haut risque rend les tests génétiques caduques.
|
© CHASSOT PG, DELABAYS A, SPAHN D Mars 2010, dernière mise à jour Août 2018
Références
- ANDERSSON C, LYNGBAEK S, NGUYEN CD, et al. Association of clopidogrel treatment with risk of mortality and cardiovascular events following myocardial infarction in patients with and without diabetes. JAMA 2012; 308:882-9
- ANGIOLILLO DJ, FERNANDEZ-ORTIZ A, BERNARDO E, et al. Variability in individual responsiveness to clopidogrel: clinical implications, management and future perspectives. J Am Coll Cardiol 2007; 49:1505-16
- ARADI D, STOREY RF, KOMOCSI A, et al. Expert position paper on the role of platelet function testing in patients undergoing percutaneous coronary intervention. Eur Heart J 2014; 35:209-15
- BATES ER, LAU WC, ANGIOLILLO DJ. Clopidogrel-drug interactions. J Am Coll Cardiol 2011; 57:1251-63
- BHATT DL, CRYER BL, CONTANT CF, et al. Clopidogrel with or without omeprazole in coronary artery disease. N Engl J Med 2010; 363:1909-17
- CATTANEO M. New P2Y12 inhibitors. Circulation 2010; 121:171-9
- CAYLA G, HULOT JS, O’CONNOR SA, et al. Clinical, angiographic, and genetic factors associated with early coronary stent thrombosis. JAMA 2011; 306:1765-74
- COOKE GE, LIU-STRATTON Y, KERKETICH AK et al. Effect of platelet antigen polymorphism on platelet inhibition by aspirin, clopidogrel, or their coombination. J Am Coll Cardiol 2006; 47:541-6
- CUISSET T, FRERE C, QUILICI J, et al. Comparison of omeprazole and pantoprazole influence on a high 150 mg clopidogrel maintenance dose. J Am Coll Cardiol 2009; 54:1149-53
- DEPTA JP, BHATT DL. Antiplatelet therapy and proton pump inhibition : cause of concern ? Curr Opin Cardiol 2012 ; 27 :642-50
- ELLIS KJ, STOUFFER GA, McLEOD HL, et al. Clopidogrel pharmacogenomics and risk of inadequate platelet inhibition: US FDA recommendations. Pharmacogenomics 2009; 10:1799-817
- GEISLER T, GAWAZ M, STEINHUBL SR, et al. Current strategies in antiplatelet therapy - Does identification of risk and adjustment of therapy contribute to more effective, personalized medicine in cardiovascular disease ? Pharmacol Ther 2010; 127:95-107
- GILARD M, ARNAUD B, CORNILY JC, et al. Influence of omeprazole on the antiplatelet action of clopidogrel associated with aspirin: The randomized, double-blind OCLA study. J Am Coll Cardiol 2008; 51:256-60
- GONZALES-CORREA JA, ARREBOLA MM, MARTIN-SALIDO E, et al. Effects of dexibuprofen on platelet function in humans: comparison with low-dose aspirin. Anesthesiology 2007; 106:218-25
- GORI AM, MARCUCCI R, MIGLIORINI A, et al. Incidence and clinical impact of dual nonresponsiveness to aspirin and clopidogrel in patients with drug-eluting stents. J Am Coll Cardiol 2008; 52:734-9
- GURBEL PA, TANTRY US. Combination antithrombotic therapies. Circulation 2010; 121:569-83
- HALL R, MAZER CD. Antiplatelet drugs: A review of their pharmacology and management in the perioperative period. Anesth Analg 2011; 112:292-318
- HO PM, MADDOX TM, WANG L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937-44
- HOLMES DR, DEHMER GJ, KAUL S, et al. ACCF/AHA clopidogrel clinical alert: Approaches to the FDA "Boxed warning". J Am Coll Cardiol 2010; 56:321-41
- HOLMES MV, PEREL P, SHAH T, et al. CYP2C19 genotype, clopidogrel metabolism, platelet function, and cardiovascular events. A systematic review and meta-analysis. JAMA 2011; 306:2704-14
- KRISHNA V, DIAMOND GA, KAUL S. Do platelet function testing and genotyping improve outcome in patients treated with antithrombotic agents ? Circulation 2012; 125:1288-303
- KULICZKOWSKI W, WITKOWSKI A, POLANSKI L, et al. Interindividual variability in the response to oral antiplatelet drugs: a position paper of the Working Group on antiplatelet drugs resistance appointed by the Section of Cardiovascular Interventions of the Polish Cardiac Society, endorsed by the Working Group on Thrombosis of the European Society of Cardiology. Eur Heart J 2009; 30:426-35
- KWOK CS, LOKE YK. Meta-analysis: the effects of proton-pump inhibitors on cardiovascular events and mortality in patients receiving clopidogrel. Aliment Pharmacol Ther 2010; 31:810-23
- LEV EI, PATEL RT, MARESH KJ, et al. Aspirin and clopidogrel drug response in patients undergoing percutaneous coronary intervention. The role of dual drug resistance. J Am Coll Cardiol 2006; 47:27-33
- MACDONALD TM, WEI L. Effect of ibuprofen on cardioprotective effect of aspirin. Lancet 2003; 361:573-4
- MARIN F, GONZALES-GONEJERO R, CAPRANZANO P, et al. Pharmacogenetics in cardiovascular antithrombotic therapy. J Am Coll Cardiol 2009; 54:1041-57
- MEGA JL, CLOSE SL, WIVIOTT SD, et al. Cytochrome P450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354-62
- MEGA JL, CLOSE SL, WIVIOTT SD, et al. Genetic variants in ABCB1, CYP2C19, and cardiovascular outcomes following treatment with clopidogrel and prasugrel. Lancet 2010; 376:1312-9
- MEGA JL, HOCHHOLZER W, FRELINGER AL, et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA 2011; 306:2221-8
- MEGA JL, SIMON T, COLLET JP, et al. Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: a meta-analysis. JAMA 2010; 304:1821-30
- MEHTA SR, YUSUF S, PETERS RJ, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527-33
- MOUKARBEL GV, BHATT DL. Antiplatelet therapy and proton pump inhibition: clinician update. Circulation 2012; 125:375-80
- OLESEN JB, GISLASON GH, CHARLOT MG, et al. Calcium-channel blockers do not alter the clinical efficacy of clopidogrel after myocardial infarction. J Am Coll Cardiol 2011; 57:409-17
- PATRONO C, ROCCA B. The future of antiplatelet therapy in cardiovascular disease. Annu Rev Med 2010; 61:49-61
- RAY WA, MURRAY KT, GRIFFIN MR, et al. Outcomes with concurrent use of clopidogrel and proton-pump inhibitors. Ann Intern Med 2010; 152:337-45
- ROBERTS JD, WELLS GA, LE MAY MR, et al. Point-of-care genetic testing for personalisation of antiplatelet treatment (RapidGene™): a prospective, randomised, prood-of-concept trial. Lancet 2012; 379:1705-11
- SANTILLI F, ROCCA B DE CRISTOFORO R, et al. Platelet cyclooxygenase inhibition by low-dose aspirin is not reflected consistently by platelet function assays: implication for aspirin “resistance”. J Am Coll Cardiol 2009; 53:667-77
- SEREBRUANY VL, CHERALA G, WILLIAMS C, et al. Association of platelet responsiveness with clopidogrel metabolism: role of compliance in the assessment of “resistance”. Am Heart J 2009; 158:925-32
- SEREBRUANY VL, STEINHUBL SR, BERGER PB, et al. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246-51
- SHULDINER AR, O’CONNEL JR, BLIDEN KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302:849-57
- SIBBING D, KOCH W, GEBHARD D, et al. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation 2010; 121:512-8
- SIBBING D SCHULZ S, BRAUN S, et al. Antiplatelet effects of clopidogrel and bleeding in patients undergoing coronary stent placement. J Thromb Haemost 2010; 8:250-6
- SILLER-MATULA JM, TRENK D, SCHRÖR K, et al. How to improve the concept of individualised antiplatelet therapy with P2Y12 receptor inhibitors – is an algorithm the answer ? Thromb Haemost 2015; 113:37-52
- SIMON T, STEG PG, GILARD M, et al. Clinical events as a function of proton pump inhibitor use, clopidogrel use, and cytochrome P450 2C19 genotype on a large nationwide cohort of acute myocardial infarction. Circulation 2011; 123:474-82
- SIMON T, VERSTUYFT C, MARY-KRAUSE M, et al. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:411-23
- WANG L, McLEOD HL, WEINSHILBOUM RM. Genomics and drug response. N Engl J Med 2011; 364:1144-53
29 Les antiplaquettaires en chirurgie cardiaque et non-cardiaque
- 29.1 Physiopathologie des thrombocytes
- 29.2 Antiplaquettaires classiques
- 29.3 Nouveaux antiplaquettaires
- 29.4 Tests d'activité plaquettaire
- 29.5 Antiplaquettaires en périopératoire
- 29.5.1 Situation
- 29.5.2 Antiplaquettaires et maladies cardiovasculaires
- 29.5.3 Arrêt des antiplaquettaires
- 29.5.4 Risque hémorragique peropératoire
- 29.5.5 Balance des risques
- 29.5.6 Recommandations pour la chirurgie non-cardiaque
- 29.5.7 Recommandations pour la chirurgie cardiaque
- 29.5.8 Antiplaquettaires et anesthésie loco-régionale (ALR)
- 29.5.9 Transfusion plaquettaire et intervention pharmacologique
- 29.6 Conclusions