




























Step 13 of 18
Anomalies de la voie éjectionnelle gauche
La sténose congénitale de la voie éjectionelle gauche peut avoir plusieurs localisations différentes :
- Sous-aortique dans la chambre de chasse du VG;
- Valvulaire proprement dite;
- Supravalvulaire à la racine de l’aorte ascendante.
A ces lésions s’ajoutent celles que l’on retrouve tout au long de l’aorte thoracique :
- Hypoplasie de l’arc aortique, arc aortique interrompu (voir Hypoplasie ventriculaire);
- Syndrome de Shone;
- Coarctation de l’aorte (voir Coarctation de l’aorte).
Obstruction de la chambre de chasse
La sténose sous-aortique fixe peut être de deux types différents. Dans le premier cas, il s'agit d'une membrane plus ou moins circulaire qui ceint la chambre de chasse depuis le septum interventriculaire jusqu’au feuillet antérieur de la valve mitrale, et occasionne l'équivalent d'une sténose aortique (Figure 14.62) (Vidéo).
Vidéo: Membrane sous-aortique dans la chambre de chasse gauche en vue long-axe; elle apparaît sous forme d'une crête fibreuse au niveau septal de la CCVD; la valve aortique est hypotrophique.
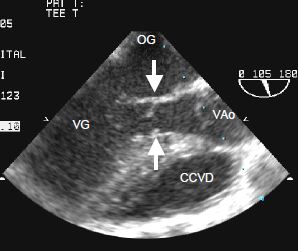

Figure 14.62 : Image ETO d’une sténose sous-aortique. Les flèches indiquent la membrane insérée sur la septum et sur la valve mitrale (feuillet antérieur).
Dans le deuxième, la CCVG est hypoplasique, longue et tubulaire; le septum est épaissi. La valve, située distalement à la membrane, est hypoplasique et incompétente dans > 50% des cas, car la sténose réduit le flux dès la naissance et le jet post-sténotique traumatise les cuspides. Ce phénomène oblige à intervenir tôt dans l’évolution de la maladie pour sauvegarder la valve aortique, soit dès que le gradient moyen est > 30 mmHg [5]. L’association avec la coarctation de l’aorte ou une CIV est fréquente.
La sténose peut aussi être dynamique et se comporter comme une cardiomyopathie obstructive (CMO). Caractérisée par une vélocité > 2.5 m/s (norme: 1.0-1.5 m/s) et un gradient maximal > 25 mmHg dans la CCVG, elle est aggravée par la stimulation sympathique β, par l'hypovolémie et par la baisse des résistances périphériques. La prise en charge implique l’arrêt des amines β, un β-bloqueur, une hypervolémie et un α-stimulant (voir Chapitre 13 Cardiomyopathie hypertrophique). Le traitement chirurgical, indiqué si le gradient est > 50 mmHg, consiste en une résection de la membrane ou une myectomie élargie de la chambre de chasse. Le risque opératoire est la création d'une CIV par résection excessive, ou un gradient persistant par résection insuffisante. L'ETO peropératoire est d'une grande importance pour diagnostiquer une CIV iatrogène ou un gradient résiduel excessif; dans 12-35% des cas, une révision chirurgicale immédiate est indiquée en fonction de cet examen [4,6].
Bicuspidie aortique
La bicuspidie aortique est rarement symptomatique chez l’enfant [2]. Elle est fréquemment associée à la coarctation de l'aorte et plus rarement à la CIV périmembraneuse. Elle se reconnaît à la présence de deux valvules au lieu de trois (voir Figures 15.49 et 15.50) (Vidéo), mais il est possible de rencontrer des cas d’unicuspidie.
Vidéo: Bicuspidie aortique dans un syndrome de Marfan; présence de deux cuspides en position antéro-postérieure.
Vidéo: Bicuspidie aortique dans un syndrome de Marfan; présence de deux cuspides en position antéro-postérieure.
Dans la bicuspidie aortique sténotique, l’indication opératoire est posée lorsque le gradient moyen est ≥ 50 mmHg, la surface ≤ 0.6 cm2/m2, le patient symptomatique ou le VG en voie de dilatation [5]. En cas d’insuffisance, l’indication est fondée sur la sévérité de l’IA et sur la dilatation du VG. Chez l’enfant, on évite de mettre en place une prothèse valvulaire, car une prothèse mécanique réclame une anticoagulation à vie et une prothèse biologique ne dure que 10 à 15 ans (voir Chapitre 11 Prothèses valvulaires). Si la valve est souple, non-calcifiée et sans insuffisance, une valvotomie percutanée par un ballon gonflable permet de lever l’obstacle et d’empêcher le remodelage ventriculaire. Le risque est de créer une insuffisance résiduelle. Le taux de succès à long terme est de 75% à 35 ans, pour une mortalité opératoire de 2.5% [1].
Si la situation ne se prête pas à une valvulotomie ni à une reconstruction, on peut transposer la valve pulmonaire en position aortique (avec réimplantation des coronaires) et mettre en place une homogreffe en position pulmonaire (opération de Ross) (Figure 14.63).
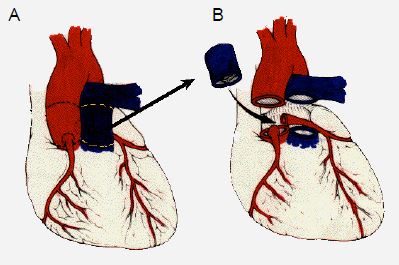
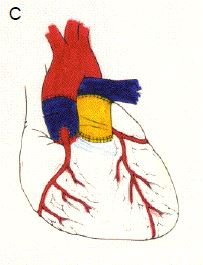
Figure 14.63. Opération de Ross. Elle consiste à réséquer la valve pulmonaire saine (A) et la valve aortique malade (B), et à implanter la valve pulmonaire du patient en position aortique ; la valve pulmonaire est remplacée par un conduit valvé entre le VD et l'AP (C) ; les coronaires sont réimplantées dans la néo-aorte.
Cette technique évite l’implantation d’une prothèse mécanique pendant la croissance et supprime l’anticoagulation. La raison de ce croisement est que le taux d’usure des homogreffes est élevé (30% à 10 ans) mais que leur dégénérescence est plus lente lorsqu’elles sont dans un système à basse pression. De plus, il est préférable d’avoir une incompétence sur la voie pulmonaire qui peut s’accommoder de grandes variations de volume, ou une sténose que l’on peut dilater par cathétérisme sans risque d’embolie systémique. Les mêmes lésions d’usure en position aortique imposeraient la mise en place d’une prothèse. Cette opération est contre-indiquée s'il existe une insuffisance pulmonaire, une bicuspidie pulmonaire ou une dysfonction ventriculaire importante. La mortalité opératoire de l’opération de Ross est de 3% et la mortalité à long-terme < 1%/an. Le taux de détérioration de l’autogreffe aortique est de 1%/an, mais celui de l’homogreffe de la voie droite est plus élevé (2-3%/an). Une insuffisance de la néovalve aortique se développe chez 30% des malades [3]. Le taux de réopération au-delà de 12 ans est de 10% pour la valve aortique et de 20-30% pour l'homogreffe pulmonaire [7].
Sténose supravalvuaire
Le rétrécissement se présente comme un diaphragme ou une déformation en sablier. Il est situé à la jonction sino-tubulaire ou au-delà des sinus de Valsalva. De ce fait, les coronaires sont en amont de la sténose et soumises au régime de l’hypertension systolique. Cette situation induit une dilatation de ces vaisseaux, accélère leur dégénérescence athéromateuse et force à intervenir tôt dans l’évolution de la maladie. L’intervention consiste en une plastie de la racine aortique avec un patch d’élargissement. Elle est indiquée si le gradient de pression moyen est ≥ 50 mmHg ou l’enfant symptomatique [5].
Anesthésie
Les contraintes de l’anesthésie sont celles d’une sténose aortique (voir Chapitre 11 Sténose aortique, Anesthésie).
- Maintien de la précharge (volume);
- Maintien de la normocardie et du rythme sinusal, esmolol en cas de tachycardie;
- Maintien de la pression de perfusion systémique par des vasoconstricteurs artériels;
- Prise en charge d’une éventuelle dysfonction du VG;
- En résumé, le malade doit être: plein – régulier - fermé.
Comme les enfants sont d'âge moyen, une induction intraveineuse est possible: étomidate (cas sévère) ou propofol (risque d'hypotension). L'anesthésie est conduite sous fentanyl (10-25 mcg/kg) ou sufentanil (0.5-2.5 mcg/kg) et sevoflurane (1-2%). Dans la sténose sus-aortique, le flux sanguin est dévié de telle manière que le flux s’engouffre dans le tronc brachiocéphalique, où la pression est plus élevée que dans la crosse distale. De ce fait, il est important de placer le cathéter artériel en position radiale gauche ou fémorale. Dans les reconstructions de la racine aortique, l’emplacement du cathéter varie selon la stratégie chirurgicale et la canulation de CEC.
Après la CEC, un gradient résiduel de 10-20 mmHg est tolérable. Les sutures aortiques étant fragiles, il est important d'éviter toute poussée hypertensive. Un bloc AV complet transitoire ou définitif est possible à cause de la proximité du nœud AV et de l'anneau aortique.
Après la CEC, un gradient résiduel de 10-20 mmHg est tolérable. Les sutures aortiques étant fragiles, il est important d'éviter toute poussée hypertensive. Un bloc AV complet transitoire ou définitif est possible à cause de la proximité du nœud AV et de l'anneau aortique.
Hypoplasie de l’arc aortique
L’arc aortique peut être sténosé ou interrompu à différents niveaux de la crosse. Cette affection est souvent accompagnée d’autres malformations cardiaques (CIV, hypoplasie du VG); elle est fréquente dans le syndrome de Di George. La circulation distale étant assurée par le canal artériel, la situation se détériore rapidement lorsque celui-ci se ferme. La reconstruction chirurgicale doit donc avoir lieu dans les premières semaines de vie; elle implique un arrêt circulatoire en hypothermie profonde pour refaçonner la crosse.
Une perfusion de prostaglandine permet de gagner quelques jours en maintenant la perméabilité du canal artériel. L’insuffisance cardiaque est bi-ventriculaire: surcharge de pression pour le VG, surcharge de pression et de volume pour le VD. De ce fait, l’anesthésie est basée sur du Fentanyl (50-75 mcg/kg) et du midazolam. Le site d’implantation du cathéter artériel est fonction de l’anatomie propre de chaque cas, mais de préférence radial droit. Lors de reconstruction de l’arc aortique, il est indiqué de placer un deuxième cathéter par voie fémorale. La sortie de CEC est caractérisée par une insuffisance ventriculaire réclamant un soutien inotrope et par une risque hémorragique élevé.
Syndrome de Shone
Le syndrome de Shone est caractérisé par une valve mitrale en parachute (voir Anomalies de la valve mitrale), un anneau sus-valvulaire mitral, une sténose sous-aortique et une coarctation de l’aorte. Les contraintes de l’anesthésie sont celles de la lésion dominante, c’est-à-dire de celle qui est la plus restrictive pour le flux gauche et la plus en amont de la série.
| Lésions sténosantes de la voie éjectionnelle gauche |
|
Trois affections congénitales :
- Sténose sous-aortique membraneuse (dysplasie valvulaire et IA fréquentes) ;
- Biscupidie valvulaire (sténose et/ou insuffisance) ;
- Sténose supra-aortique.
La prise en charge anesthésique est celle d’une sténose aortique : Plein – Régulier - Fermé
|
© BETTEX D, BOEGLI Y, CHASSOT PG, Juin 2008, dernière mise à jour Mai 2018
Références
- DETTER C, FISCHLEIN T, FELDMEIER C, et al. Aortic valvotomy for congenital valvular aortic stenosis. Ann Thorac Surg 2001; 715:1564-71
- FRIEDMAN WF. Aortic stenosis. In: EMMANOULIDES GC, ed. Moss and Adam's heart disease in infants, children and adolescents including fetus and young adult. Baltimore, Williams & Wilkins 1995, p 1087
- FRIGIOLA A, RANUCCI M, CARLUCCI C, et al. The Ross procedure in adults: long-term follow-up and echocardiographic changes leading to pulmonary autograft reoperation. Ann Thorac Surg 2008; 86:482-9
- ROSENFELD HM, GENTLES TL, WERNKOVSKY G, et al. Utility of intraoperative echocardiography in the assessment of residual cardiac defects. Ped Cardiol 1998; 19:346-51
- SILVERSIDES CK, KIESS M, BEAUCHESNE L, et al. Canadian Cardiovascular Society 2009 Consensus Conference on the management of adults with congenital heart disease: Outflow tract obstruction, coarctation of the aorta, tetralogy of Fallot, Ebstein anomaly and Marfan’s syndrome. Can J Cardiol 2010; 26:e80-e97
- STEVENSON JG, SORENSEN GK, GARTMAN DM, et al. Left ventricular outflow tract obstruction: An indication for intraoperative transesophageal echocardiography. J Am Soc Echocardiogr 1993; 6:525-35
- TAKKENBERG JJM, KLIEVERIK LMA, SCHOOF PH, et al. The Ross procedure: a systematic review and meta-analysis. Circulation 2009; 119:222-8
14. Anesthésie pour la chirurgie cardiaque pédiatrique
- 14.1 Introduction
- 14.2 Physiopathologie
- 14.3 Stratégies hémodynamiques
- 14.3.1 Classification
- 14.3.2 Shunt G → D et débit pulmonaire élevé
- 14.3.3 Hypertension pulmonaire pédiatrique
- 14.3.4 Shunt droit → gauche et débit pulmonaire abaissé
- 14.3.5 Shunt droit → gauche cyanogène et débit systémique abaissé
- 14.3.6 Shunt cyanogène mixte
- 14.3.7 Cardiopathies sans shunt : obstructions et valvulopathies
- 14.3.8 Options thérapeutiques chez le nouveau-né
- 14.3.9 Pharmacothérapie
- 14.4 Techniques d'anesthésie
- 14.5 La CEC chez l'enfant
- 14.6 Approche par pathologie
- 14.6.1 Introduction
- 14.6.2 Repères anatomiques
- 14.6.3 Retours veineux anormaux
- 14.6.4 Communication interauriculaire (CIA)
- 14.6.5 Canal atrio-ventriculaire (CAV)
- 14.6.6 Maladie d'Ebstein
- 14.6.7 Anomalies des valves auriculo-ventriculaires
- 14.6.8 Communication interventriculaire (CIV)
- 14.6.9 Hypoplasie ventriculaire
- 14.6.10 Tétralogie de Fallot
- 14.6.11 Ventricule droit à double issue
- 14.6.12 Atrésie pulmonaire
- 14.6.13 Anomalies de la voie éjectionnelle gauche
- 14.6.14 Transposition des gros vaisseaux
- 14.6.15 Truncus arteriosus
- 14.6.16 Coarctation de l'aorte
- 14.6.17 Anomalies artérielles
- 14.6.18 Transplantation cardiaque
- 14.7 Conclusions