




























Step 9 of 17
Période suivant la CEC
Décanulations
La canule veineuse de l'OD est enlevée en premier. La canule aortique est enlevée avant ou après la première demi-dose de protamine, selon les centres, mais jamais après l'injection de toute la dose, car les risques de thrombose et d'embolie artérielle seraient trop grands. La paroi de l'aorte est souvent fragile, particulièrement chez les malades âgés et artériosclérotiques. Il est important que la pression artérielle soit basse à ce moment-là (PAM 50-60 mmHg) pour éviter une déchirure lorsque la bourse est serrée. L'ablation des canules de l'OD améliore le retour veineux.
Il est fréquent que le chirurgien soulève le cœur pour contrôler l'hémostase et les anastomoses latérales ou postérieures. Cette manipulation bloque momentanément le retour veineux et induit des arythmies ventriculaires polymorphes; la pression artérielle s'effondre. Le seul traitement est le repositionnement du cœur. Il n'y a pas lieu d'utiliser des anti-arythmiques ou des agents cardiopresseurs, car leur pic d'activité coïnciderait avec la décharge sympathique qui suit la manœuvre et conduirait à une poussée hypertensive majeure. La vitesse de récupération de la pression artérielle au repositionnement du cœur est un bon indice de la performance ventriculaire: lorsque celle-ci est médiocre, cette durée est prolongée.
Après la CEC
Le relâchement de l'écarteur sternal est le premier geste qui s'accompagne d'une stimulation douloureuse; il est suivit de la coagulation du sternum, de la pose des fils métalliques et de la fermeture de la cage thoracique. Une dose adéquate d'opiacé est nécessaire pour bloquer la réponse sympathique et empécher le réveil. Malgré des problèmes hémodynamiques souvent difficiles, il est capital de s'assurer que le patient soit correctement endormi. La meilleure manière d’assurer l’anesthésie est un débit continu d'halogéné sur le ventilateur ou une perfusion de propofol par voie centrale; elle peut aussi être garantie par des injections itératives ou une perfusion de midazolam. Le N2O est contre-indiqué à cause du risque d'hypertension pulmonaire et d'aggravation des embolies gazeuse. Il n'y a pas de "vitesse de croisière" pour le débit des catécholamines dans le postopératoire immédiat, car la situation est très rapidement évolutive.
La curarisation n'est pas nécessaire pour fermer une sternotomie car la musculature thoracique ne contribue pas à l'écartement du sternum. Par contre, les frissons sont une indication à un curare parce qu'ils augmentent la VO2 jusqu'à 400%; le cœur en phase de récupération n'est pas à même de fournir cet excès de travail [3]. S'il n'a pas été suffisamment réchauffé en CEC (T° rectale > 35°, T° oesophagienne > 36°), le corps reviendra lentement à la normothermie aux dépens d'une élévation du DC à un moment où ce dernier est limité, ce qui est également une situation défavorable. D'autre part, la vasoconstriction hypothermique persistante augmente les RAS, donc la postcharge et le travail du VG; elle séquestre des territoires musculaires qui restent peu vascularisés, mais qui représentent des réserves de sang froid remis en circulation au fur et à mesure qu'ils reprennent une perfusion normale.
L'acide tranexamique (30 mg/kg) est administré après la normalisation de l'ACT par la protamine. Malgré ces deux mesures, les antiplaquettaires préopératoires, la CEC et la réaction inflammatoire systémique conjuguent leurs effets dans un tableau complexe d'hypocoagulabilité. Dans cette situation, le thromboélastogramme (ROTEM™) est d'une très grande utilité pour sélectionner la meilleure thérapeutique: plaquettes, desmopressine, fibrinogène, complexe prothombinique, facteur VIII, facteur XIII, facteur VIIa, etc. L’utilisation systématique du thromboélastogramme et des tests de fonction plaquettaire à la sortie de pompe est un progrès incontestable dans la gestion globale des transfusions et des dérivés sanguins [6,32].
La prise en charge hémodynamique comporte plusieurs volets (Figure 4.24 et Tableau 4.15).
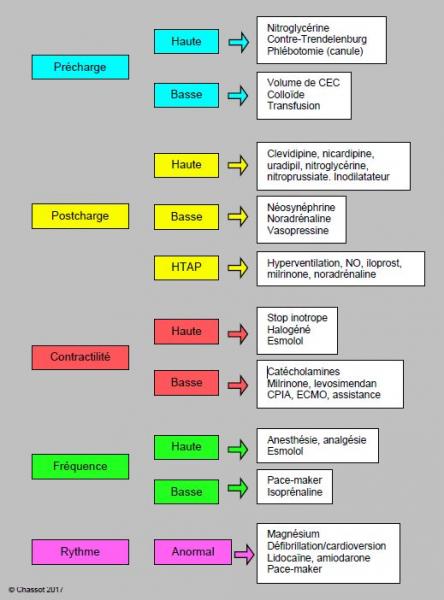
Figure 4.24: prise en charge hémodynamique dans la période qui suit la CEC. Les cinq paramètres essentiels sont constamment ajustés pour obtenir la meilleure performance en terme de pression artérielle (cathéter artériel systémique, cathéter de Swan-Ganz), de débit cardiaque (thermodilution, PiCCO) et de fonctionnement myocardique (ETO).

- Précharge: la dysfonction diastolique post-CEC augmente les pressions (PVC et PAPO) pour le même volume de remplissage; toutefois, la distension des ventricules est la catastrophe à éviter. L'ETO permet de suivre adéquatement le volume des cavités cardiaques. L'inspection visuelle à travers la sternotomie montre le VD et l'OD; le VD est dilaté s'il déborde en hauteur le bord de l'incision péricardique au niveau du diaphragme.
- Postcharge: elle est mesurée par les RAS et les RAP; elle est réglée par la profondeur de l'anesthésie, les vasodilatateurs systémiques (isoflurane, phentolamine, nitroprussiate, nitroglycérine) ou pulmonaires (prostaglandines, milrinone, NO•), et les vasoconstricteurs (phényléphrine, nor-adrénaline, vasopressine). L’hypotension artérielle signe aussi bien une baisse des RAS qu’une dysfonction du VG ; un vasopresseur seul améliore le chiffre de la PA mais aggrave la défaillance gauche.
- Contractilité: le débit cardiaque visé est un IC ≥ 2.2 L/min/m2; les catécholamines sont choisies en fonction des RAS, des RVP et de la prédominance des récepteurs α ou β dans le myocarde. En effet, l'insuffisance ventriculaire et la CEC diminuent la population de récepteurs β, ce qui amoindrit l'efficacité des amines qui agissent par l'intermédiaire de ce récepteur. L'activité de la milrinone et du levosimandan est indépendante des récepteurs catécholaminergiques.
- Fréquence cardiaque: idéalement située entre 70 et 80 batt/min pour assurer un DC satisfaisant, elle est réglée en fonction des contraintes hémodynamiques propres à la cardiopathie. La dysfonction diastolique rend le débit particulièrement sensible au variations de fréquence: il baisse aussi bien lorsque la FC augmente (diastole trop courte pour le remplissage lent des défauts de relaxation) que lorsque la FC baisse (ventricule trop rigide pour augmenter son Vtd et compenser la bradycardie).
- Rythme: le rythme sinusal procure le meilleur débit, particulièrement en cas de dysfonction diastolique où la contraction auriculaire assure 25-50% du remplissage ventriculaire. La présence d'une onde P sur l'ECG garantit un rythme sinusal électrique, mais ne signifie pas que la contraction auriculaire soit efficace. Seule la visualisation du flux mitral à l'ETO permet de constater la contribution réelle du flux A au Vtd du VG. Avant la fermeture du péricarde, on s'assure que les électrodes ventriculaire et auriculaire entraînent correctement, que les seuils sont bas et que le pace-maker est fonctionnel.
L'ischémie myocardique résiduelle après pontages aorto-coronariens se manifeste par un sus-décalage ST persistant sur l'ECG et des altérations de la cinétique segmentaire non régressive à l'ETO. Elle a plusieurs origines possibles.
- Revascularisation coronarienne incomplète.
- Sidération (non-reprise fonctionnelle malgré le rétablissement de la circulation), phénomène de no-reflow (absence de perfusion périphérique alors que le vaisseau épicardique débite correctement).
- Lésion de reperfusion: accumulation de Ca2+ ionisé dans le sarcoplasme, libération de radicaux libres (peroxydes) qui lèsent les lipoprotéines, œdème cellulaire; sur l'ECG, on voit des ondes T inversées et pointues.
- Spasme des coronaires ou des pontages artériels sur une surstimulation sympathique β, une alcalose respiratoire, une stimulation plaquettaire (libération de thromboxane) ou une lésion de l'endothélium (sécrétion d'endothéline et répression du NO•).
- Distension et subocclusion du pontage mammaire par un volume courant excessif ou une PEEP exagérée.
- Embolie athéromateuse intracoronarienne (l’embolie gazeuse n’est pas persistante).
- Défaut sur une anastomose, thrombose coronarienne aiguë.
Plusieurs mesures sont à prendre si les symptômes ischémiques persistent > 15 minutes.
- Augmentation de la pression de perfusion (nor-adrénaline) pour PAM > 80 mmHg;
- Mesure du flux dans les pontages au moyen d'une sonde Doppler;
- Reprise chirurgicale et réfection des anastomoses;
- Perfusion de nitroglycérine (50 mcg/min); certains centres le font systématiquement pour toutes les interventions de revascularisation coronarienne;
- En cas de spasme: diltiazem (Dilzem®) 0.1 mg/kg/heure en perfusion sans dose de charge; clevidipine (Cleviprex®) 1-2 mg/h en perfusion, à doubler toutes les 3-5 minutes jusqu’à l’effet désiré (dose maximale : 32 mg/h) [13].
- Administration intracoronarienne de papavérine ou d'adénosine.
Une sténose dynamique de la chambre de chasse du VG survient facilement après une CEC lorsque s’associent quatre phénomènes :
- Hypertrophie ventriculaire gauche (HVG) concentrique;
- Diminution de la taille de la cavité ventriculaire en systole (hyovolémie, stimulation β) ;
- Stimulation β-adrénergique excessive ;
- Baisse de la postcharge (vasoplégie, remplacement d’une valve aortique sténosée par une prothèse).
Dans ces conditions, la zone de coaptation de la valve mitrale est déplacée antérieurement et la partie distale du feuillet antérieur, destabilisée, est poussée vers la chambre de chasse où elle est aspirée par effet Venturi pendant l’éjection ; ce phénomène est appelé systolic anterior motion (SAM) (voir Chapitre 13, Cardiomyopathie hypertrophique). Le feuillet antérieur vient alors occlure partiellement la CCVG et le débit systolique baisse brusquement. C’est l’effet CMO, appelé ainsi par analogie au mécanisme de la cardiomyopathie obstructive (Vidéos et Figures 4.25 et 4.26) [10].
Vidéo: subobstruction méso-systolique de la chambre de chasse du VG par la partie distale du feuillet mitral antérieur (SAM: systolic anterior motion), typique de l'effet CMO.
Vidéo: subobstruction méso-systolique de la chambre de chasse du VG par la partie distale du feuillet mitral antérieur (SAM: systolic anterior motion), typique de l'effet CMO.
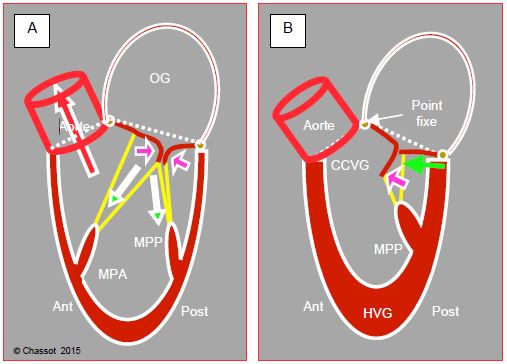
Figure 4.25 : Contraction du VG. A : Situation normale. L’augmentation de la pression intra-ventriculaire en systole applique les deux feuillets de la valve mitrale l’un contre l’autre (flèches violettes) ; la contraction de la paroi et des muscles papillaires (MPA : muscles papillaire antérieur, MPP : muscle papillaire postérieur) tend les cordages et maintient la valve mitrale dans son plan d’occlusion (flèches vertes). Ceci constitue le squelette interne du ventricule. Le sang est expulsé dans la chambre de chasse du VG (CCVG). La contraction débute à l’apex et se termine dans la chambre de chasse. B : Rétrécissement de la cavité ventriculaire. Si la course de la paroi postérieure et du MPP est trop longue (surstimulation β, vasoplégie) ou si le ventricule est trop petit (hypovolémie, hypertrophie concentrique), la paroi postérieure se déplace vers l’avant (flèche verte) ; le point de coaptation de la valve mitrale est déplacé antérieurement vers l’aorte et se rapproche de la CCVG. Le feuillet antérieur est alors repoussé vers la CCVG lorsque la pression intraventriculaire augmente pendant la systole (flèche violette). Traitillé blanc : angle mitro-aortique. L’angle mitro-aortique est un point fixe puisqu’il est situé au niveau du trigone fibreux, qui est le centre de gravité mécanique du cœur sur lequel sont ancrées les valves mitrale, aortique et tricuspide. Si l’anneau mitral se rétrécit, le seul déplacement possible est celui de sa partie postérieure, qui est essentiellement musculaire, et qui se retrouve alors tirée vers l’avant.
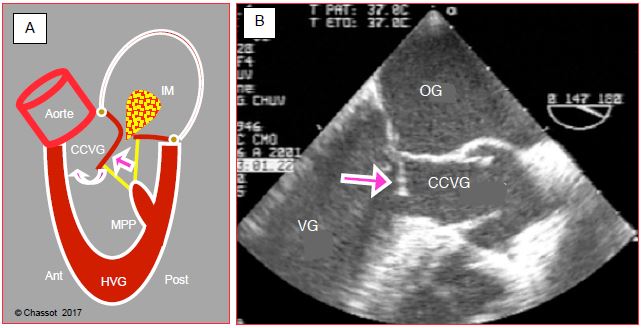
Figure 4.26 : Effet CMO en cas de restriction de la cavité ventriculaire gauche et d’avancement excessif de la paroi postérieure. A : la pression de la phase de contraction isovolumétrique pousse le feuillet vers l’avant en direction de la CCVG. Dès que l’éjection commence, la partie distale de ce feuillet peut être aspirée dans la CCVG par effet Venturi et induire un phénomène appelé systolic anterior motion (SAM). Le feuillet antérieur vient alors occlure partiellement la CCVG et le débit systolique baisse brusquement. C’est l’effet CMO, appelé ainsi par analogie avec le mécanisme de la cardiomyopathie obstructive. La réouverture de la valve mitrale en mésosystole provoque une insuffisance mitrale méso-télésystolique (IM). B : illustration échocardiographique transoesophagienne (vue mi-œsophage long-axe 120°) ; la flèche indique la coudure du feuillet mitral antérieur (SAM) qui vient obstruer la chambre de chasse gauche (CCVG). Les cuspides de la valve aortique sont ouvertes.
Cliniquement, l'effet CMO se caractérise par une hypotension artérielle et un bas débit cardiaque qui s’aggravent avec les catécholamines. C’est une situation que seule l’échocardiographie permet de diagnostiquer, mais qui est particulièrement traître parce que le réflexe normal face à un bas DC est d’augmenter la stimulation adrénergique. Or le seul traitement est une augmentation de la volémie (remplissage), une vasoconstriction (phényléphrine, noradrénaline) et une diminution de l'effet β (arrêt des catécholamines, esmolol). En cas d’hypertrophie du VD, le même phénomène peut se produire dans la chambre de chasse droite [3].
La fermeture du péricarde et du sternum comprime le cœur et augmente la pression intrathoracique; c'est l'équivalent d'une tamponnade momentanée avec accentuation des interactions cardio-respiratoires. Cette manœuvre est mal supportée lorsque le patient est hypovolémique ou lorsque le cœur est dilaté par une défaillance. Il est nécessaire d'augmenter la précharge (remplissage) et le soutien inotrope (catécholamines) pour maintenir la pression artérielle et le débit cardiaque. Si un choc cardiogène s'installe, on rouvre le sternum et le péricarde. Lors de défaillance droite ou de pathologie pulmonaire, il est judicieux de ne refermer ni le péricarde ni le sternum afin d'éviter cette compression du VD. L'ETO permet de contrôler l'absence de tamponnade par des caillots, qui serait une indication à la réouverture immédiate pour drainage, et l'absence de modification de la cinétique segmentaire, qui signerait la coudure ou la compression d'un pontage. Si le sternum est laissé ouvert, la plaie est protégée par une pièce de tissu synthétique (GoreTex™, Dacron™) cousue à la peau, et le sternum sera refermé 48-72 heures plus tard.
Afin d’améliorer le confort et l’analgésie postopératoires des cas dont on ne prévoit pas une extubation accélérée, il est judicieux d’administrer de la morphine i.v. (0.1-0.2 mg/kg) en fin de CEC ou pendant la fermeture, à un moment où le status cardiocirculatoire est stable. En vue d'un réveil rapide, une perfusion de dexmédétomidine (0.2-1.5 mcg/kg/heure) agit comme sédation et potentialisation de l'analgésie [14,23]; elle est particulièrement indiquée après une anesthésie aux halogénés. Dans le cas d'un maintien par une perfusion de propofol (1.5-2.5 mg/kg/h), celle-ci est continuée jusqu'aux soins intensifs. Le BIS et le SPI n'étant pas des garanties contre l'éveil et la douleur peropératoires, il est prudent de maintenir les patients sous-curarisés de manière à pouvoir juger de l'adéquation de l'anesthésie par l'apparition de mouvements respiratoires ou somatiques et à pouvoir procéder plus rapidement à l'extubation. L'examen ETO final est réalisé après la fermeture du sternum, de manière à déceler d'éventuelles complications: tamponnade, hémothorax, pneumothorax, compression ou défaillance ventriculaire, etc.
Dysfonctions organiques post-CEC
La CEC est liée à une série de dysfonctions postopératoires, bien qu'elle n'en soit pas la cause unique [27].
- Insuffisance rénale transitoire dans 22% des cas, nécessitant une dialyse dans 3.5% (1-6.5%) [5,9]. Les facteurs de risque sont : dysfonction rénale préopératoire (créatinine > 200 μmol/L, clairance < 50 mL/min), transfusion sanguine (> 2 unités), produit de contraste (< 5 jours préop), hypotension/hypovolémie/bas débit peropératoire, hypothermie profonde, longue durée de la CEC; le débit urinaire en CEC n'a pas de valeur prédictive (voir Chapitre 23 Complications rénales) [21].
- Oedème pulmonaire non cardiogénique suite à l'augmentation de la perméabilité capillaire, à la baisse du surfactant, à la ventilation contrôlée, à l'accumulation liquidienne péricapillaire, aux atélectasies et aux transfusions (TRALI: transfusion-related acute lung injury). L'incidence des complications pulmonaires est de 10-25% des cas; un SDRA survient dans 1.3 - 1.7% des CEC (voir Chapitre 23 Pneumopathies postopératoires) [17,30].
- Hypoxie secondaire:
- Oedème pulmonaire cardiogénique (défaillance gauche, valvulopathie mitrale);
- Pneumothorax, hémothorax;
- Obstruction bronchique (sécrétions, sang, déplacement du tube endotrachéal);
- Atélectasie;
- Effet shunt (inhibition de la vasoconstriction pulmonaire hypoxique par les vasodilatateurs artériels, foramen ovale perméable en cas de surcharge droite).
- Rupture d'une artère bronchique par le gonflement intempestif du ballonnet de Swan-Ganz alors que le cathéter est encore en position bloquée.
- Séquelles neurologiques [22,24,31]:
- Type I (ischémie transitoire, accident vasculaire cérébral, coma; incidence 1-5%), dues principalement à des embols (athéromatose de l'aorte ascendante, débris dans les aspirations, air) ;
- Type II (neuropsychologiques diffuses, délire; incidence > 30%) plutôt liée à l'hypoxie cérébrale et à l'hypotension en CEC;
- Facteurs de risque: anamnèse d'AVC, athéromatose aortique, maladie polyvasculaire, diabète, âge > 70 ans. Les séquelles neurologiques sont liées en premier lieu à la charge embolique, alors que l'hypotension joue un rôle plus modeste chez l'adulte.
- Diminution de la perfusion hépato-splanchnique: risque d'endotoxémie et d’insuffisance hépatique ; la clairance du lactate est diminuée de 30% par la CEC [19].
- Syndrome inflammatoire systémique: voir ci-dessous.
Syndrome inflammatoire systémique
Une intervention de chirurgie cardiaque déclenche une vigoureuse réponse de défense de la part de l'organisme, caractérisée par un relargage massif de cytokines pro-inflammatoires (TNF-α, IL-1α, IL-1β, IL-6, IL-8) et anti-inflammatoires (IL-10, TNF 1-2SR). A l'image de la réaction inflammatoire locale en cas de dommage tissulaire, la réaction systémique produit une vasodilatation, un exsudat capillaire et de la fièvre. Par ses liens avec la voie du complément, elle active la cascade de la coagulation, la fibrinolyse et la libération de bradykinine, d'histamine, de leukotriènes ainsi que d'entotoxines issues des bactéries du tube digetif, mais elle stimule le système immunitaire et diminue la vulnérabilité aux infections [16]. Les endotoxines, les kinines, le système du complément et les cytokines sont les éléments humoraux de la réaction; ces médiateurs activent les neutrophiles et les cellules endothéliales. L'adhésion des leucocytes à ces dernières est l'étape initiale de la réaction; elle est déclenchée par l'expression de molécules spécifiques à la surface des deux types de cellules; les leucocytes peuvent alors migrer dans l'espace extravasculaire, où ils libèrent leurs toxines (protéases et radicaux libres) qui détruisent les pathogènes mais endommagent les tissus voisins [8,18,26,28]. Le syndrome inflammatoire systémique (Systemic Inflammatory Reaction Syndrome ou SIRS) est l'aboutissement commun de cette pléthore de modifications hématologiques, sériques, immunes, protéiques et toxiques. Il est déclenché par un série de phénomènes: l'intrusion chirurgicale, le contact du sang avec les surfaces étrangères du circuit de CEC et avec l'air, l'héparinisation, les aspirations de cardiotomie, l'hypothermie, l'ischémie, l'exclusion des poumons, les variations de flux, de pression et de débit (voir Chapitre 7, Syndrome inflammatoire systémique). Environ 20% des patients à bas risque développent des complications liées au SIRS (Figure 4.27) [12].
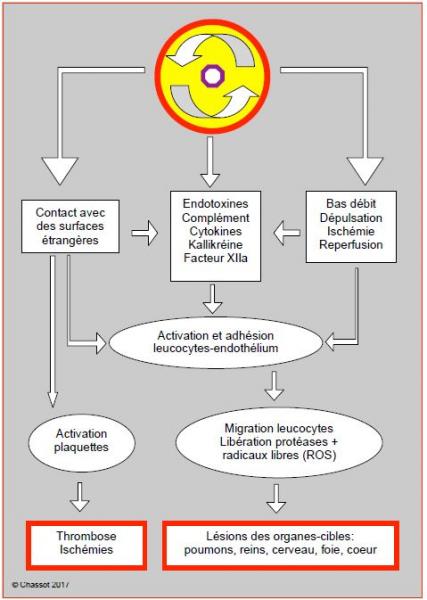
Figure 4.27: Représentation schématique des mécanismes mis en jeu dans la genèse du syndrome inflammatoire systémique par la CEC [d'après références 8 et 18].
Sans en dresser une liste exhaustive, on peut citer un certain nombre d'éléments qui entrent en jeu dans la genèse du SIRS, qui se caractérise cliniquement par l'accumulation liquidienne interstitielle, l'oedème cérébral, la péjoration des échanges gazeux et des fonctions rénales, des troubles de la coagulation et différentes complications organiques postopératoires (insuffisance polyorganique) [26,28].
- Les effets des de cytokines pro-inflammatoires (TNF-α, interleukines IL-1α, IL-1β, IL-6 et IL-8) sont partiellement compensés par la stimulation des cytokines anti-inflammatoires (IL-10, TNF-1SR, TNF-2SR) dont le but est de contenir l'ampleur de la réaction.
- L'activation de la cascade du complément par la voie alternative (C3a et C5a) stimule les leucocytes qui adhèrent aux parois endothéliales par le truchement de molécules spéciales, les sélectines et les intégrines. Ils sécrètent des cytokines, des enzymes protéolytiques et des radicaux libres, et provoquent l'adhésion plaquettaire. L'activation de la cascade du complément est aussi responsable de la mise en route de la chaîne intrinsèque de la coagulation [29].
- L'activation de la cascade de coagulation; le contact avec des surfaces étrangères active le facteur XII (Hageman factor) en facteur XIIa en présence de prékallikréine et de kininogène; celui-ci active à son tour le facteur XI et la cascade de coagulation se met en marche (voir Figure 8.1) [18].
- La libération de kallikréine par le contact avec les surfaces étrangères engendre celle de la bradykinine; cette dernière augmente la perméabilité capillaire, contracte la musculature lisse, et provoque une hypotension. La bradykinine est directement en cause dans la genèse de l'oedème cérébral [7].
- La libération d'endothéline, qui est un puissant vasoconstricteur, est stimulée par la CEC; elle est liée à l'hypertension pulmonaire. La thromboxane A2 sécrétée par les leucocytes activés est aussi un vasoconstricteur et un stimulant de l'aggrégation plaquettaire.
- Les radicaux libres (ROS: Reactive Oxygen Species) contiennent un nombre impair d'électrons sur leur orbite externe: peroxide (O2•-), H2O2, hydroxyl (•OH); ils se forment lors d'activation leucocytaire ou lors de reperfusion après ischémie. Normalement réduits par des antioxydants naturels (superoxide dismutase, catalase, glutathion, vitamine E), ils débordent dans le liquide extracellulaire lorsqu'ils sont produits en masse et attaquent les phospholipides des membranes, le DNA, et les protéines [11].
- L'élimination des cytokines est un phénomène primairement rénal; ceci implique un effet de rétro-action réciproque entre le syndrome inflammatoire et la dysfonction rénale [1].
- La durée de CEC, la profondeur de l'hypothermie et le degré d'hémodilution ont tous été évoqués comme facteurs aggravants, mais ils ne paraissent avoir qu'un rôle secondaire dans la genèse du SIRS [8]. Les lésions mécaniques de la pompe, de l'oxygénateur et des filtres, le contact du sang avec les surfaces étrangères (circuits) et avec l'air (aspirations, réservoir veineux), sont les éléments déclenchants principaux. Plus de 50% des neutrophiles sont séquestrés dans les poumons durant le réchauffement; leur dégranulation contribue aux dommages cellulaires pulmonaires [17].
- La libération d'endotoxines par les bactéries Gram-négatives qui hantent le tube digestif est liée à la baisse de débit splanchnique en CEC et en hypothermie; le flux dans la muqueuse gastrique, par exemple, est diminué jusqu'à 60% [20].
- La libération de cytokines pro-inflammatoire démarre dès la sternotomie [15]. D'autre part, la chirurgie à coeur battant est associée à une réduction, mais pas à une disparition, des taux postopératoires de marqueurs de la réaction inflammatoire [2]. Le circuit de CEC est donc un facteur déclenchant majeur, mais il n'est pas le seul responsable de la réaction inflammatoire. Celle-ci est un corollaire de tout acte chirurgical.
Les effets hémodynamiques du SIRS dépendent largement de l'équilibre entre la libération des médiateurs pro-inflammatoires et celle des médiateurs anti-inflammatoires [12]. Certaines populations affichent une réaction pro-inflammatoire dominante, telles les personnes âgées ou celles qui souffrent de dysfonction ventriculaire gauche. Le syndrome inflammatoire est donc une réponse physiologique à une agression tissulaire, destinée à promouvoir les défenses infectieuses et la cicatrisation. Elle est normalement auto-limitée par ses éléments anti-inflammatoires, mais peut échapper à ce contrôle lorsque le stimulus est trop important, telle une intervention majeure couplée à un contact prolongé avec des surfaces étrangères.
Syndrome vasoplégique
La vasoplégie est un état de choc distributif caractérisé par des RAS effondrées (500-800 dynes•s•cm-5), une hypotension sévère (PAM < 50 mmHg) et une hypoperfusion des organes malgré un débit cardiaque conservé ou élevé. Elle survient dans 5-25% des cas après CEC [4,25]. L’étiopathogénie est certainement multifactorielle, mais on décèle un certain nombre de facteurs de risque [25].
- Médication préopératoire: inhibiteurs de l'enzyme de conversion, béta-bloqueurs;
- Etat inflammatoire accentué comme dans certaines comorbidités ou dans l'insuffisance ventriculaire gauche (FE basse);
- CEC: longue durée, libération d'endotoxines, lésion d'ischémie-reperfusion (radicaux libres), activation du complément, déclenchement du syndrome inflammatoire systémique (cytokines, NO, prostaglandines, thromboxane, etc);
- Post-CEC: sécrétion élevée de NO•, production abaissée de vasopressine;
- Vasodilatation de la protamine.
Le syndrome vasoplégique survient aussi après chirurgie à cœur battant (sans CEC), mais trois fois plus rarement. Il nécessite un traitement vascoconstricteur intense : nor-adrénaline, adrénaline, vasopressine, bleu de méthylène (voir Vasopresseurs). Les vasopresseurs non-catécholaminergiques tendent à diminuer le besoin en catécholamine et le taux d'insufisance rénale postopératoire, mais rien de prouve qu'ils diminuent la mortalité [25]. Comme la chute des RAS facilite l'éjection du VG, le traitement vasopresseur doit s'accompagner d'une surveillance échocardiographique de la tolérance du ventricule à l'augmentation de sa postcharge.
| Période suivant la CEC |
|
Le relâchement de l’écarteur sternal est la première stimulation douloureuse après la CEC (approfondir l’anesthésie et l’analgésie) ; la fermeture du sternum ne réclame pas de curarisation. Par contre, le curare est indiqué pour stopper les frissons (↑ VO2).
La fermeture du péricarde et du sternum est l’équivalent d’une tamponnade qui demande une augmentation momentanée de la précharge et éventuellement un support inotrope du VD. En cas de défaillance droite, il est recommandé de laisser le sternum ouvert.
Le débit cardiaque et la performance ventriculaire sont réglés en ajustant la précharge (volume de la CEC, colloïdes, transfusions), la postcharge (vasopresseur ou vasodilatateur), la contractilité (catécholamines), la fréquence et le rythme (antiarythmiques, pace-maker). Les besoins en support inotrope sont très variables après une intervention cardiaque et doivent être réévalués en permanence. L’hypotension artérielle signe aussi bien une baisse des RAS qu’une dysfonction du VG ; un vasopresseur seul améliore le chiffre de la PA mais aggrave la dysfonction ventriculaire.
Une ischémie myocardique se manifeste par une surélévation ST (ECG) et des altérations de la cinétique segmentaire (ETO). Ses causes sont une embolie gazeuse (lésion passagère), une sidération, une revascularisation incomplète, un spasme, une thrombose, une lésion de reperfusion, un défaut d’anastomose ou une distension du greffon mammaire par une ventilation excessive. Traitement : augmenter la PAM (> 80 mmHg), nitroglycérine, diltiazem, reprise chirurgicale, CPIA.
L’intervention cardiaque et la CEC sont à l’origine d’un syndrome inflammatoire systémique majeur.
|
© CHASSOT PG, BETTEX D, MARCUCCI C, Septembre 2010, dernière mise à jour, Décembre 2018
Références
- BAKER R, ALLEN S, ARMSTRONG MA, et al. Role of the kidney in perioperative inflammatory responses. Br J Anaesth 2002; 88:330-4
- CZERNY M, BAUMER H, KILO J, et al. Inflammatory response and myocardial injury following coronary artery bypass grafting with or without cardiopulmonary bypass. Eur J Cardiothorac Surg 2000; 17: 737-42
- DENAULT AY, DESCHAMPS A, COUTURE P. Intraoperative hemodynamic instability during and after separation from cardiopulmonary bypass. Semin Cardiothorac Vasc Anesth 2010; 14:165-82
- FISCHER GW, LEVIN MA. Vasoplegia during cardiac surgery: current concepts and management. Semin Thorac Cardiovasc Surg 2010; 22:140-4
- GAFFNEY AM, SLADEN RN. Acute kidney injury in cardiac surgery. Curr Opin Anaesthesiol 2015; 28:50-9
- GÖRLINGER K, DIRKMANN D, HANKE AA ; et al. First-line therapy with coagulation factor concentrates combined with point-of-care coagulation testing is associated with decrease allogeneic blood transfusion in cardiovascular surgery. Anesthesiology 2011 ; 115 : 1179-91
- HARRIS DN, OATRIDGE A, DOB D, et al. Cerebral swelling after normothermic cardipulmonary bypass. Anesthesiology 1998; 88:340-7
- HENNEIN HA. Inflammation after cardiopulmonary bypass: therapy for the postpump syndrome. Semin Cardiothorac Vasc Anesth 2001; 5:236-55
- HU J, CHEN R, LIU S, et al. Global incidence and outcomes of adult patients with kidney injury after cardiac surgery: a systematic review and meta-analysis. J Cardiothorac Vasc Anesth 2016; 30:82-9
- JAIN P, PATEL PA, FABBRO M. Hypertrophic cardiomyopathy and left ventricular outflow tract obstruction: expecting the unexpected. J Cardiothorac Vasc Anesth 2018; 32:467-77
- KRISHNADASAN B, HAMPTON CR, GRISCAVAGE-ENNIS J, et al. Molecular mechanisms of neurologic injury following cardiopulmonary bypass. Semin Cardiothorac Vasc Anesth 2002; 6:43-53
- LAFFEY JG, BOYLAN JF, CHENG DC. The systemic inflammatory response to cardiac surgery. Anesthesiology 2002; 97:215-52
- LIU H, FOX CJ, ZHANG S, KAYE AD. Cardiovascular pharmacology: an update. Anesthesiology Clin 2010; 28:723-38
- LIU H, FUHAI JI, PENG K, et al. Sedation after cardiac surgery: is one drug better than another ? Anesth Analg 2017; 124:1061-70
- McBRIDE WT, ARMSTRONG MA, CROCKARD AD, et al. Cytokine balance and immunosuppressive changes at cardiac surgery: contrasting response between patients and isolated CPB circuits. Br J Anaesth 1995; 75:724-33
- McBRIDE WT, McBRIDE ER. The inflammation response to cardiothoracic surgery. In: ALSTON RP, MYLES P, RANUCCI M, eds. Oxford Textbook of Cardiothoracic anesthesia. Oxford: Oxford University Press, 2015, 61-7
- MESSENT M, SULLIVAN K, KEOGH BF, et al. Adult respiratory distress syndrome following cardiopulmonary bypass: incidence and prediction. Anaesthesia 1992; 47: 267-8
- MILLER BE, LEVY JH. The inflammatory response to cardiopulmonary bypass. J Cardiothorac Vasc Anesth 1997; 11:355-66
- MUSTAFA I, ROTH H, HANAFIAH A, et al. Effect of cardiopulmonary bypass on lactate metabolism. Intensive Care Med 2003; 29:1279-85
- OHRI SK, BOWLES CW, MATHIE RT, et al. Effect of cardiopulmonary bypass perfusion protocols on gut tissue oxygenation and blood flow. Ann Thorac Surg 1997; 64:163-8
- O'NEAL JB, SHAW AD, BILLINGS FT. Acute kidney injury following cardiac surgery: current understanding and future directions. Crit Care 2016; 20:187
- ROACH GW, KANCHUGER M, MORA MANGANO C, et al. Adverse cerebral outcomes after coronary bypass surgery. N Engl J Med 1996; 335:1857-64
- SCHNABEL A, MEYER-FRIEBEM C, REICHL S, et al. Is intraoperative dexmedetomidine a new option for postoperative pain treatment ? A meta-analysis of randomized controlled trials. Pain 2013; 154: 1140-9
- SELNES OA, GOTTESMAN RF, GREGA MA, et al. Cognitive and neurologic outcomes after coronary artery bypass surgery. N Engl J Med 2012; 366:250-7
- SHAEFI S, MITTEL A, KLICK J, et al. Vasoplegia after cardiovascular procedures – Pathophysiology and targeted therapy. J Cardiothorac Vasc Anesth 2018; 32:1013-22
- SHERWOOD ER, TOLIVER-KINSKY T. Mechanisms of the inflammatory response. Best Pract Res Clin Anaesthesiol 2004; 18:385-405
- SHROYER AL, COOMBS LP, PETERSON ED, et al. The Society of Thoracic Surgeons: 30-day operative mortality and morbidity risk models. Ann Thorac Surg 2003; 75:1856-64
- SNIECINSKI RM, LEVY JH. The inflammatory response to cardiopulmnary bypass. In: MONGERO LB, BECK JR (eds). On bypass. Advanced perfusion techniques. Totowa (NJ, USA): Humana Press 2010, 125-140
- STAHL GL, SHERMAN SK, SMITH PK, LEVY JH. Complement activation and cardiac surgery : a novel target for improving outcomes. Anesth Analg 2012 ; 115 :759-71
- UBBEN JFH, LANCE MD, BUHRE WF, SCHREIBER JU. Clinical strategies to prevent pulmonary complications in cardiac surgery: an overview. J Cardiothorac Vasc Anesth 2015; 29:481-90
- VAN WERMESKERKEN GK, LARDENOYE JW, HILL SE, et al. Intraoperative physiologic variables and outcome in cardiac surgery: Part II. Neurologic outcome. Ann Thorac Surg 2000; 69:1077-83
- WEBER CF, GÖRLINGER K, MEININGER D. Point-of-care testing : a prospective randomized clinical trial of efficacy in coagulopathic cardiac surgery patients. Anesthesiology 2012 ; 117 : 531-47
04. Spécificités de l'anesthésie pour la chirurgie cardiaque
- 4.1 Remarques générales
- 4.2 Effets hémodynamiques des agents d'anesthésie
- 4.3 Conduite de l'anesthésie en chirurgie cardiaque
- 4.3.1 Prémédication
- 4.3.2 Choix de la technique d'anesthésie
- 4.3.3 Induction
- 4.3.4 Ventilation
- 4.3.5 Période précédant la circulation extra-corporelle
- 4.3.6 Anticoagulation pour la CEC
- 4.3.7 L'anesthésie pendant la circulation extra-corporelle
- 4.3.8 Sevrage de la CEC
- 4.3.9 Période suivant la CEC
- 4.3.10 Insuffisance ventriculaire post-CEC
- 4.3.11 Arythmies post-CEC
- 4.3.12 Réveil et extubation
- 4.3.13 Circuit rapide (fast-track)
- 4.3.14 Besoins liquidiens
- 4.3.15 Epargne sanguine et transfusions
- 4.3.16 Contrôle métabolique
- 4.3.17 Situations particulières
- 4.4 Anesthésie loco-régionale
- 4.5 Médicaments cardiovasculaires peropératoires
- 4.6 Conclusions