




























Step 2 of 3
Activation plaquettaire
Lorsque survient une lésion endothéliale, les plaquettes entrent en contact avec le collagène, avec le facteur von Willebrand, et éventuellement avec les lipides d’une plaque athéromateuse instable ; cette dernière est constituée d’un amas de lipides, de macrophages et d’éléments inflammatoires recouverts d’une fine capsule. Lors de cette rupture endothéliale, la rencontre entre les structures sous-endothéliales et les thrombocytes circulants active spécifiquement le récepteur GP Ib/V/IX et provoque une dégranulation plaquettaire qui libère de la thromboxane A2 (TXA2), de l’adénosine-diphosphate (ADP) et de la thrombine. Ces substances stimulent à leur tour les récepteurs correspondants de chaque plaquette et ceux des thrombocytes voisins ; elles amplifient ainsi la réaction par recrutement de nouveaux éléments (Figure 29.1) [3]. Les plaquettes activées changent de forme et développent des spicules typiques.
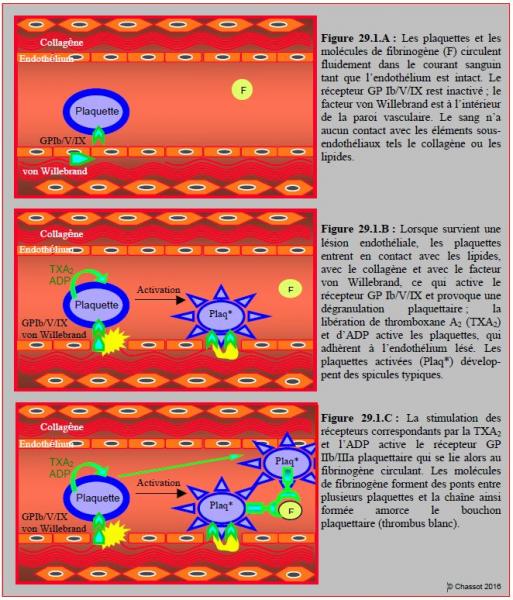
Figure 29.1 : Activation des plaquettes et formation d’un thrombus plaquettaire (thrombus blanc) lors de lésion endothéliale mettant à nu les lipides et le collagène normalement recouverts d’endothélium : phase initiale (29.1.B) et phase d’extension (29.1.C). Les agents antiplaquettaires (aspirine, clopidogrel, etc) agissent en bloquant la phase d’activation des plaquettes. Plaq* : plaquette activée.
La stimulation des récepteurs plaquettaires par la TXA2, l’ADP et la thrombine transmet un signal intracellulaire qui conduit à une hausse du Ca2+ ionisé sarcoplasmique et à une baisse de la production d’AMP cyclique (cAMP) ; ce phénomène active le complexe GP IIb/IIIa situé à la surface des plaquettes. Une fois activé, ce dernier se lie au fibrinogène circulant. Cette liaison forme des ponts entre plusieurs plaquettes et devient le ciment qui agglutine les thrombocytes entre eux. La chaîne ainsi formée amorce le bouchon plaquettaire (thrombus blanc).
Lorsqu’elles sont stimulées, les plaquettes libèrent des agents activateurs de la coagulation (TXA2, ADP, sérotonine, thrombine) dont certains sont aussi des vasoconstricteurs locaux (TXA2, sérotonine), alors que l’endothélium sécrète des substances qui freinent l’activité plaquettaire et ont un effet vasodilatateur : le NO• (baisse du Ca2+ ionisé intracellulaire), la prostacycline PGI2 (augmentation du cAMP, modulation de la réponse au TXA2) et l’ecto-ADPase (suppression de la phase de recrutement plaquettaire) (Figure 29.2).
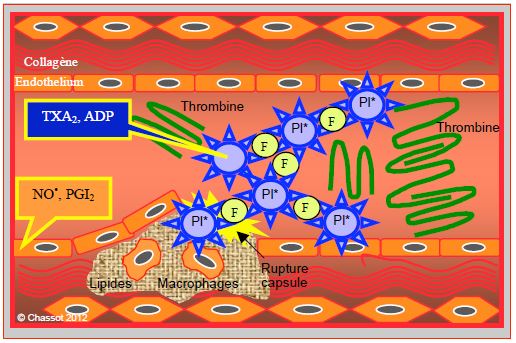
Figure 29.2 : Une plaque instable est constituée d’un amas de lipides, de macrophages et d’éléments inflammatoires recouverts d’une fine capsule. Lorsque celle-ci vient à se rompre, les structures sous-endothéliales entrent en contact avec les éléments circulants : les plaquettes sont activées, s’aggluttinent autour des molécules de fibrinogène et déclenchent l’adhésion de molécules de thrombine. Les plaquettes sécrètent des agents activateurs de la coagulation (TXA2, ADP) et vasoconstricteurs (TXA2), alors que l’endothélium sécrète des substances qui freinent l’activité plaquettaire et sont vasodilatatrices (NO•, PGI2).
La situation peut évoluer de deux manières [2] :
- Prédominance des agents vasodilatateurs et inhibiteurs des plaquettes : résolution spontanée du thrombus ; la circulation est rétablie mais la cicatrisation de la plaque accroît progressivement sa taille.
- Prédominance des agents vasoconstricteurs et stimulateurs des plaquettes comme le stress, la fumée ou l’inflammation : recrutement massif de thrombocytes et de facteurs de coagulation ; le thrombus devient occlusif et provoque une ischémie ou une nécrose distale.
L’activité endocrine opposée des plaquettes et de l’endothélium maintient un équilibre dynamique au niveau artériolaire. Mais certaines situations sont accompagnées d’une excitabilité plaquettaire exagérée (production excessive de ligand CD-40 et de radicaux libres) : obésité, tabagisme, hyperlipidémie, hypercholestérolémie, hypertension artérielle, vieillissement, diabète, insuffisance rénale [1].
| Activation plaquettaire |
|
L’endothélium normal protège les éléments circulants de tout contact avec les structures de la paroi vasculaire (collagène, lipides, facteurs tissulaires). Une lésion endothéliale (brèche vasculaire, rupture de plaque instable) stimule les plaquettes circulantes par activation du récepteur GP Ib/V/IX via le collagène et le facteur von Willebrand (FvW). Les plaquettes sécrètent alors de la thromboxane A2 (TXA2), de l’ADP, de la thrombine et de la sérotonine, qui vont activer les récepteurs correspondants situés sur la plaquette (amplification) et sur ses voisines (recrutement). Ces différents récepteurs contribuent à l’élévation du Ca2+ ionisé intracellulaire et à la baisse de l’AMP cyclique (cAMP). Le point d’aboutissement est la stimulation de la glycoprotéine (GP) IIb/IIIa qui se lie au fibrinogène et constitue ainsi des amas plaquettaires (thrombus blanc). La plaquette est ancrée à la paroi vasculaire par liaison au FvW. Le déclenchement de la cascade de la coagulation accumule de la thrombine et de la fibrine (thrombus rouge).
|
© CHASSOT PG, DELABAYS A, SPAHN D Mars 2010, dernière mise à jour Août 2018
Références
- DAVI G, PATRONO C. Platelet activation and atherothrombosis. N Engl J Med 2007; 357:2482-94
- FALK E, SHAH PK, FUSTER V. Coronary plaque disruption. Circulation 1955; 92:657-71
- SAKHUJA R, YEH RW, BHATT DL. Antiplatelet agents in acute coronary syndromes. Curr Probl Cardiol 2010; 35:123-70
29 Les antiplaquettaires en chirurgie cardiaque et non-cardiaque
- 29.1 Physiopathologie des thrombocytes
- 29.2 Antiplaquettaires classiques
- 29.3 Nouveaux antiplaquettaires
- 29.4 Tests d'activité plaquettaire
- 29.5 Antiplaquettaires en périopératoire
- 29.5.1 Situation
- 29.5.2 Antiplaquettaires et maladies cardiovasculaires
- 29.5.3 Arrêt des antiplaquettaires
- 29.5.4 Risque hémorragique peropératoire
- 29.5.5 Balance des risques
- 29.5.6 Recommandations pour la chirurgie non-cardiaque
- 29.5.7 Recommandations pour la chirurgie cardiaque
- 29.5.8 Antiplaquettaires et anesthésie loco-régionale (ALR)
- 29.5.9 Transfusion plaquettaire et intervention pharmacologique
- 29.6 Conclusions