




























Step 4 of 9
Pulmonary hypertension
Pathophysiology
Pulmonary hypertension (PHT) is defined as mean resting PAP > 25 mmHg or pulmonary vascular resistance (PVR) > 300 dynes•cm•s-5 (see Chapter 12 Pulmonary hypertension) [17,18]. Pulmonary arterial hypertension (PAH) observed in 5-10% of congenital heart disease patients is more specifically an increase in pre-capillary pressure. PHT is split into 5 categories [17,23,37].
Pulmonary hypertension (PHT) is defined as mean resting PAP > 25 mmHg or pulmonary vascular resistance (PVR) > 300 dynes•cm•s-5 (see Chapter 12 Pulmonary hypertension) [17,18]. Pulmonary arterial hypertension (PAH) observed in 5-10% of congenital heart disease patients is more specifically an increase in pre-capillary pressure. PHT is split into 5 categories [17,23,37].
- 1 - Pulmonary arterial hypertension (PAH): PAH due to congenital heart diseases, primary idiopathic PAH, drug/infection/age-related PAH.
- 1’ - Veno-occlusive pulmonary disease.
- 1’’ - Persistent PAH in neonates (2: 1,000 babies).
- 2 - Post-capillary pulmonary hypertension (PHT) (LVEDP > 18 mmHg, PAWP > 15 mmHg, but PVR < 3 Wood Units and transpulmonary gradient < 12 mmHg): systolic failure or restrictive diastolic failure of the LV, mitral valvular disease.
- 3 - PAH associated with alveolar hypoxia: COPD, emphysema, ARDS, sleep apnoea syndrome, high-altitude hypoxia.
- 4 - PAH related to chronic pulmonary thromboembolic disease.
- 5 - Unexplained multi-factorial PAH.
In groups 1, 3, 4 and 5, PAH is pre-capillary. LAP and PAWP are normal (PAWP < 15 mmHg), and the transpulmonary gradient (TPG = mPAP - LAP, or mPAP - PAWP) is > 12 mmHg. PVR is high (> 240 dynes•s•cm-5 or > 3 Wood U). PHT related to left heart pathologies (group 2) is primarily post-capillary: high LAP and PAWP (> 15 mmHg), low PVR and transpulmonary gradient (TPG < 12 mmHg). If pulmonary arterial pressure is removed from the equation for calculating vascular resistance, this gives: PAP ≈ LAP + (CO • PVR). PA pressure is therefore dependent on three factors: LA pressure (left-sided stasis, post-capillary PHT), pulmonary blood flow (left-to-right shunting, hyperkinetic PHT) and pulmonary vascular resistance (pulmonary arterial hypertension, pre-capillary PAH) (Figure 15.65) [27,31].
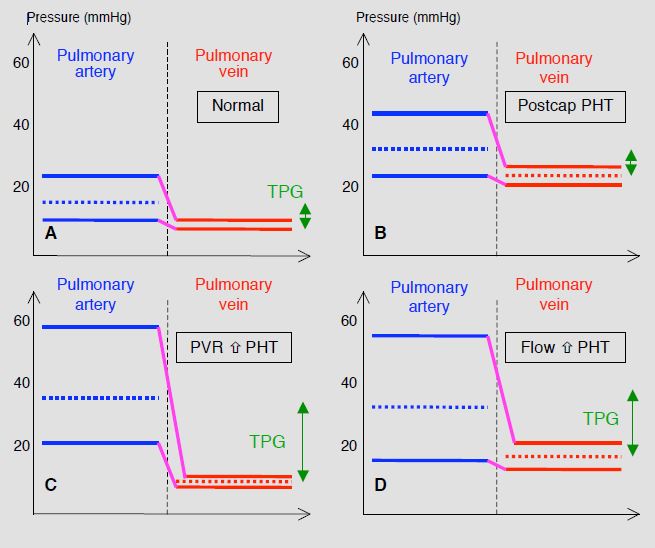
Figure 15.65: Schematic classification of the various mechanisms of pulmonary hypertension (PHT) [31]. A: normal situation – the transpulmonary gradient (TPG, green double arrow) is < 12 mmHg. B: post-capillary or passive PHT – venous pressure is high, resulting in a secondary increase in PAP. However, the transpulmonary gradient remains normal. C: pre-capillary or resistance PHT – the primary mechanism is an increase in PVR. Venous pressure is normal and the TPG is very high (> 12 mmHg). D: hyperkinetic PHT secondary to an increase in pulmonary blood flow (L-to-R shunt). PAP is high, but venous pressure is slightly higher than normal levels, moderately reducing the increase in the TPG. Since PVR increases secondarily to the flow overload, PHT in grown-up congenital heart patients is a combination of C and D.
The most common cause of PAH in congenital heart patients is a non-restrictive L-to-R shunt. It occurs in 50% of cases of VSD and patent ductus arteriosus, where both volume and pressure overload are present, but in only 10% of cases of ASD, where volume overload only is concerned. It appears in infancy in cases of VSD, but only in adulthood in cases of ASD [30]. Vascular wall shear stress caused by excessive pulmonary blood flow causes an expansion of the smooth muscle in peripheral vessels that are normally not muscular. At this stage, pressure remains at the upper limit of normal levels [34]. The steady rise in pressure is associated with hypertrophy of the media of more proximal arteries combined with progressive intimal proliferation and fibrosis followed by thickening of the tunica externa, an inflammatory reaction and disseminated thrombosis [32] (Figure 15.66).
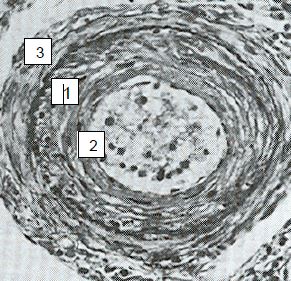
Figure 15.66: Anatomopathological image of a pulmonary arteriole in Eisenmenger’s syndrome. 1: medial hypertrophy. 2: intimal proliferation. 3: fibrosis [32]. The wall of such an artery can no longer be collapsed by airway pressure under mechanical ventilation.
Finally, the number of distal vessels decreases – PAH is fixed and irreversible. This is Eisenmenger's syndrome, characterised by mean PAP > 50 mmHg at rest, PVR > 800 dynes•cm•s-5 and bidirectional shunting (L-to-R shunt + R-to-L component) combined with cyanosis [17]. Inversion of the L-to-R shunt is the main cause of sudden cyanosis onset in adult congenital heart disease patients [13]. PAH irreversibility is demonstrated by testing vasoreactivity to NO, whereby potential reversibility is defined as a reduction of mean PAP by > 10 mmHg and a reduction of mean PAP to ≤ 40 mmHg, without reducing cardiac output [5,18].
The mass of the two ventricles is equal during intrauterine life; the RV retains its foetal structure if it is exposed to a high afterload from birth and hypertrophies in parallel to the development of the LV [8]. It resists the increase in afterload more effectively than if this occurs secondarily in adulthood. Although surgical correction is no longer possible if the PVR/SVR ratio is higher than 0.7, outcomes for grown-up congenital heart patients are better than those of patients suffering from primary, drug-induced or thromboembolic PAH [6,22,24].
Figure 15.65: Schematic classification of the various mechanisms of pulmonary hypertension (PHT) [31]. A: normal situation – the transpulmonary gradient (TPG, green double arrow) is < 12 mmHg. B: post-capillary or passive PHT – venous pressure is high, resulting in a secondary increase in PAP. However, the transpulmonary gradient remains normal. C: pre-capillary or resistance PHT – the primary mechanism is an increase in PVR. Venous pressure is normal and the TPG is very high (> 12 mmHg). D: hyperkinetic PHT secondary to an increase in pulmonary blood flow (L-to-R shunt). PAP is high, but venous pressure is slightly higher than normal levels, moderately reducing the increase in the TPG. Since PVR increases secondarily to the flow overload, PHT in grown-up congenital heart patients is a combination of C and D.
The most common cause of PAH in congenital heart patients is a non-restrictive L-to-R shunt. It occurs in 50% of cases of VSD and patent ductus arteriosus, where both volume and pressure overload are present, but in only 10% of cases of ASD, where volume overload only is concerned. It appears in infancy in cases of VSD, but only in adulthood in cases of ASD [30]. Vascular wall shear stress caused by excessive pulmonary blood flow causes an expansion of the smooth muscle in peripheral vessels that are normally not muscular. At this stage, pressure remains at the upper limit of normal levels [34]. The steady rise in pressure is associated with hypertrophy of the media of more proximal arteries combined with progressive intimal proliferation and fibrosis followed by thickening of the tunica externa, an inflammatory reaction and disseminated thrombosis [32] (Figure 15.66).
Figure 15.66: Anatomopathological image of a pulmonary arteriole in Eisenmenger’s syndrome. 1: medial hypertrophy. 2: intimal proliferation. 3: fibrosis [32]. The wall of such an artery can no longer be collapsed by airway pressure under mechanical ventilation.
Finally, the number of distal vessels decreases – PAH is fixed and irreversible. This is Eisenmenger's syndrome, characterised by mean PAP > 50 mmHg at rest, PVR > 800 dynes•cm•s-5 and bidirectional shunting (L-to-R shunt + R-to-L component) combined with cyanosis [17]. Inversion of the L-to-R shunt is the main cause of sudden cyanosis onset in adult congenital heart disease patients [13]. PAH irreversibility is demonstrated by testing vasoreactivity to NO, whereby potential reversibility is defined as a reduction of mean PAP by > 10 mmHg and a reduction of mean PAP to ≤ 40 mmHg, without reducing cardiac output [5,18].
The mass of the two ventricles is equal during intrauterine life; the RV retains its foetal structure if it is exposed to a high afterload from birth and hypertrophies in parallel to the development of the LV [8]. It resists the increase in afterload more effectively than if this occurs secondarily in adulthood. Although surgical correction is no longer possible if the PVR/SVR ratio is higher than 0.7, outcomes for grown-up congenital heart patients are better than those of patients suffering from primary, drug-induced or thromboembolic PAH [6,22,24].
- ASD: 90% of 40-year survival;
- Eisenmenger's syndrome: 80% of 10-year survival;
- Primary idiopathic PAH: 66% of 5-year survival;
- Thromboembolic disease: 35% of 5-year survival.
The prognosis for PAH is much more closely linked to right-sided function than to the PAP value. It is poor if the RV fails and dilates. Indeed, PAH is dependent on the RV’s ability to chronically generate high pulmonary pressure. Once the RV fails, PAP tends to fall again, while the haemodynamic situation worsens and SVR continues to rise (Figure 15.67) [21].
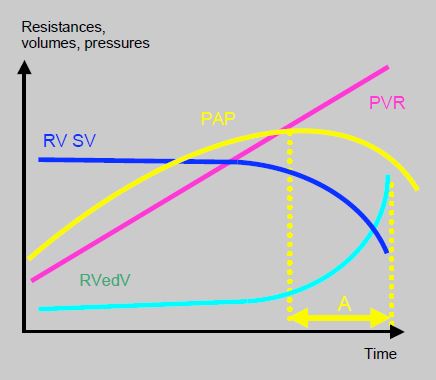
Figure 15.67: Relationship between the RV and pulmonary circulation. Overview of the relationship between pulmonary vascular resistance (PVR), RV stroke volume (RVSV), RV end-diastolic volume (RVedV), and pulmonary arterial pressure (PAP). PAH is dependent on the RV’s ability to chronically generate high pulmonary pressure. Once the RV fails, PAP tends to fall again, while the haemodynamic situation worsens and SVR continues to rise. In time period A (yellow), measured PAP is lower than previously, not because the situation is improving, but due to a deterioration in RV function. The clinical severity and prognosis of the disease are therefore determined more by right ventricular function than by the PAP value in itself [21].
For anaesthetists, pulmonary hypertension in grown-up adult congenital heart patients exhibits certain pathophysiological characteristics that are common to all forms of PAH, with the exception of the first form listed below, which is specific to congenitals [4,7,11,15,18,27].
Figure 15.67: Relationship between the RV and pulmonary circulation. Overview of the relationship between pulmonary vascular resistance (PVR), RV stroke volume (RVSV), RV end-diastolic volume (RVedV), and pulmonary arterial pressure (PAP). PAH is dependent on the RV’s ability to chronically generate high pulmonary pressure. Once the RV fails, PAP tends to fall again, while the haemodynamic situation worsens and SVR continues to rise. In time period A (yellow), measured PAP is lower than previously, not because the situation is improving, but due to a deterioration in RV function. The clinical severity and prognosis of the disease are therefore determined more by right ventricular function than by the PAP value in itself [21].
For anaesthetists, pulmonary hypertension in grown-up adult congenital heart patients exhibits certain pathophysiological characteristics that are common to all forms of PAH, with the exception of the first form listed below, which is specific to congenitals [4,7,11,15,18,27].
- In L-to-R shunts, reduced systemic pressure due to hypovolaemia or systemic vasodilation exacerbates the shunt's R-to-L component, reduces pulmonary blood flow, and exacerbates cyanosis.
- Pulmonary blood flow is reduced and fixed. Any compliance with variations in blood flow and circulating volume is lost. It is impossible to increase Qp if there is an increased O2 requirement: patients become desaturated at the slightest effort.
- Systemic hypotension lowers coronary perfusion pressure, increases the interventricular septum’s shift to the left, and threatens subendocardial perfusion of the RV.
- The more hypertrophied the RV is, the more it behaves like the LV. Its Frank-Starling curve is steeper and its blood flow becomes dependent on its preload. In such cases, hypovolaemia causes a fall in pulmonary blood flow and hypoxaemia.
- During diastole, pressure from the hypertrophied and overloaded RV is higher than that from the LV. The interventricular septum bulges into the LV (Bernheim effect) reducing its diastolic filling. It is usually possible to replace the septum in its physiological position by increasing left-sided afterload (systemic arterial vasoconstriction).
- Pulmonary vascular reactivity is preserved despite the thickening of vessel walls – hypercapnia, acidosis and hypoxaemia further increase PVR while hyperventilation reduces it [7,9]. Anaesthetists must therefore avoid any procedures that might exacerbate PVR [11]:
- Hypoventilation: hypercapnia, hypoxaemia, atelectasis;
- High intrathoracic pressure;
- Acidosis;
- Hypothermia;
- Anaemia (Hb < 100 g/L);
- Sympathetic stimulation, stress, pain.
PAH is one of the key indicators of surgical risk in congenital heart disease patients. Only lifesaving procedures may be considered for these patients [10].
Treatment of pulmonary hypertension
Anaesthetists must take all possible steps to reduce pulmonary blood pressure (see Chapter 12 PAH treatment). It is essential for patients to be deeply anaesthetised (stress-free anaesthesia, high-dose fentanyl, sufentanil or remifentanil) and remain normothermic. The first stage in acute treatment of PAH involves ventilation, since respiratory alkalosis, gas inhalation and substance nebulisation are the only techniques capable of lowering pulmonary arterial pressure without causing systemic hypotension. Inhalation offers the double benefit of not causing hypotension or impeding hypoxic pulmonary vasoconstriction in non-ventilated areas, since substances are only distributed to the ventilated alveoli. Consequently, hypoxaemia is not exacerbated (Table 15.6) [7,11,17,25, 27,38].
Treatment of pulmonary hypertension
Anaesthetists must take all possible steps to reduce pulmonary blood pressure (see Chapter 12 PAH treatment). It is essential for patients to be deeply anaesthetised (stress-free anaesthesia, high-dose fentanyl, sufentanil or remifentanil) and remain normothermic. The first stage in acute treatment of PAH involves ventilation, since respiratory alkalosis, gas inhalation and substance nebulisation are the only techniques capable of lowering pulmonary arterial pressure without causing systemic hypotension. Inhalation offers the double benefit of not causing hypotension or impeding hypoxic pulmonary vasoconstriction in non-ventilated areas, since substances are only distributed to the ventilated alveoli. Consequently, hypoxaemia is not exacerbated (Table 15.6) [7,11,17,25, 27,38].
- Hyperventilation: hypocapnia (PaCO2 30-35 mm Hg) and alkalosis (pH 7.5) have a pulmonary vasodilator effect without affecting the systemic vascular bed. FiO2 must be high, ventilation pressure must be low and the tidal volume must be similar to the FRC (6-8 mL/kg.). pH rather than pCO2 regulates the pulmonary vascular tone. PVR reactivity to acidosis and alkalosis is increased in cases of hypoxia and pulmonary hypertension [7]. Ideally, the ventilator should be set to ensure a minimal mean ventilation pressure (6-10 mmHg) for ETCO2 of 25-30 mmHg.
- Administering O2 (glasses, mask) outside the operating theatre is beneficial, especially in severe cases of erythrocytosis (Ht > 55%).
This is followed by drug treatment. Some substances can be nebulised into the respiratory circuit and therefore have no systemic impact [38,39].
- NO: 10 – 30 ppm in the ventilator's inspiratory circuit. No systemic effect – the NO is immediately inactivated by its bond with haemoglobin, but it impairs platelet function. In cases of pulmonary hypertension or situations where PAP needs to be kept low (e.g. Fontan), NO must be administered on induction and/or resumption of ventilation after CPB, before any hypertensive crises occur. Weaning from NO may be difficult due to the rebound effect on PAH, which may be mitigated by administering sildenafil, prostacyclin or milrinone. If PVR does not fall within 30 minutes of administering NO•, this indicates that the substance is ineffective and treatment must be discontinued [3].
- Prostacyclins: iloprost as a nasal spray (Ilomedin®, 10-20 mcg in 20 min) to be repeated every 3-4 hours (30-minute half-life), or in continuous nebulisation (0.2-0.3 mL/min of a 10-20 mcg/mL solution). In acute treatment, prostacyclins have an additive effect with NO•. Prostacyclins reduce platelet aggregability.
- Phosphodiesterase-3 inhibitors – when nebulised rather than infused, milrinone exhibits less of a systemic hypotensive effect and greater pulmonary efficacy. Nebulisation of a 1 mg/mL solution at 0.2-0.3 mL/min for 10-20 minutes, i.e. 50-80 mcg/kg in 10 min. It is less expensive than NO and prostaglandins, and does not cause any rebound effects when discontinued. Iloprost and milrinone are also easier to use than NO•.
- Nitroglycerin: nebulised at a rate of 2.5 mcg/kg/min – less effective than the above agents.
There is a set of substances capable of acutely lowering PAP by intravenous route. However, all these pulmonary vasodilators exhibit varying degrees of systemic hypotensive effects [25,27].
- Prostaglandins: by stimulating adenylyl cyclase, they increase cAMP levels and prompt vasodilation. When used in chronic and acute treatment, they have an additive effect with NO [35, 36].
- Epoprostenol (Flolan®) as an infusion (2-5 ng/kg/min) – very short half-life (4-6 min);
- Iloprost (Ilomedin®) 10-20 mcg over 20 min – duration of action: 60 minutes;
- Treprostinil (Remodulin®) subcutaneous or IV (1.25-2.5 ng/kg/min); 3-hour half-life.
- Phosphodiesterase-5 inhibitor – sildenafil (Revatio®, 3 x 10 mg/d IV); it is used concurrently with NO and prostaglandins. Particularly effective in combination with L-arginine [19,29]. PDE5 inhibitors are the only class of pulmonary vasodilators that have a beneficial effect on PHT linked to left heart pathologies [20].
- Magnesium: Mg2+ (5-10 mmol) exhibits some degree of pulmonary dilator effect similar to a calcium channel blocker.
- Arginine: through its transformation into citrulline, arginine chloride (15 mg/kg/min) provides the substrate required for producing NO. It is probably indicated after CPB as this lowers arginine levels.
- Citrulline: this NO• precursor is capable of reducing PAH in cases of paediatric congenital heart disease (9 mg/kg/hour infusion). Only limited data are currently available.
- Nitroglycerin (0.5-5 mcg/kg/min): slight pulmonary vasodilator effect in addition to reducing RV end-diastolic volume and improving right-sided congestive failure at high CVP. Risk of systemic hypotension by reducing preload. It is only indicated for congenital heart disease patients in the event of RV congestive failure (severe tricuspid or pulmonary insufficiency).
- Nesiritide (initial dose 2 mcg/kg, infusion 0.01-0.03 mcg/kg/min): this BNP derivative lowers PVR and improves haemodynamics in the event of right-sided failure secondary to PAH. Its clinical impact is probably minimal.
- Calcium channel blockers: contraindicated in congenital heart disease patients since systemic vasodilation in excess of pulmonary vasodilation can intensify the cyanotic effect of a bidirectional shunt [18].
- It is essential to maintain systemic MAP above PAP (MAP/mPAP > 3) with an arterial vasoconstrictor (norepinephrin or vasopressin) for three reasons:
- To reduce the shunt's R-to-L component if necessary.
- To correct the position of the interventricular septum (convex in the RV) and restore assistance to right-sided ejection provided by LV contraction. An increase in left-sided afterload is required to cause the septum to bulge into the RV during systole.
- To maintain coronary perfusion of the RV as it can only rely on the systolic component of coronary blood flow if PAP is close to MAP [38].

RV performance is essential for ensuring haemodynamic balance. For right-sided defects, several positive inotropic agents offer pulmonary vasodilator activity in addition to their cardiostimulatory effects and are therefore the preferred choice for patients with pulmonary hypertension.
- Dobutamine (Dobutrex®): by increasing cAMP (β1 stimulation), it induces significant pulmonary vasodilation.
- Inodilators: phosphodiesterase-3 inhibitors reduce PAP and PVR, and increase EF through their positive inotropic effect – amrinone (Inocor®), milrinone (Corotrop® 0.5 mcg/kg/min).
- Epinephrin + milrinone is generally the most effective combination as it balances inotropic, pulmonary vasodilator, and systemic vasoconstrictor effects.
- Levosimendan (initial dose 6 mcg/kg, infusion 0.05-0.2 mcg/kg/min): this reduces PAP (right-sided afterload) and increases ventricular contractility by sensitising troponin to Ca2+. It is the only inotropic agent that reduces mortality [33].
- Isoprenaline (Isuprel®): very effective, but causes severe tachycardia and systemic hypotension (β1 + β2 stimulation).
- Perioperatively, opening or non-closure of the pericardium and sternum to decompress the RV and reduce ventricular interdependence.
- Ventricular assistance: ECMO, RV ventricular assist device (Impella™, etc.).
The haemodynamic balance of congenital heart disease patients with PAH is very delicate. It is therefore essential to adopt a proactive approach and initiate a hypotensive treatment as soon as PAP begins to rise (mPAP > 0.5 MAP). An increase in CVP and/or a reduction in stroke volume are warning signs indicating that right-sided function must be supported pharmacologically. Moreover, by monitoring the RV by TEE, it is possible to initiate inotropic support as soon as RV performance deteriorates or its volume increases.
Patients with PAH usually receive chronic treatment with pulmonary vasodilators. This treatment must be strictly maintained perioperatively [17,18].
Patients with PAH usually receive chronic treatment with pulmonary vasodilators. This treatment must be strictly maintained perioperatively [17,18].
- Endothelin receptor antagonists: Bosentan (Tracleer®, 125-250 mg 2 x/d), sitaxsentan (100 mg/d);
- Phosphodiesterase-5 inhibitors – sildenafil (Viagra®, Revatio®, 3 x 20 mg/d oral), tadalafil, vardenafil;
- Prostaglandins: beraprost, iloprost;
- cGMP stimulant: riociguat (Adempas®);
- Losartan (angiotensin II receptor antagonist) at a rate of 50-100 mg/d.
- Calcium channel blockers are ineffective or even dangerous in congenital heart disease patients.
Since the pulmonary vessels have virtually no α1 receptors, arteriolar vasodilators (sodium nitroprusside, phentolamine) mainly have a systemic effect and are not useful for selective treatment of PAH. Their effect is catastrophic in subjects with a R-to-L shunt or whose pulmonary blood flow is supplied from the systemic blood flow (L-to-R Blalock-Taussig shunt, aortopulmonary collaterals). Given this small population of α receptors, norepinephrin and phenylephrine have a negligible pulmonary vasoconstrictor effect. While norepinephrin increases the PVR/SVR ratio, vasopressin reduces it, since the systemic vasoconstrictor effect of vassopressin is not associated with an increase in PVR, given the predominance of vasodilator V2 receptors over vasoconstrictor V1 receptors in the pulmonary vascular bed [26]. Pulmonary vasodilators are contraindicated for PHT secondary to left heart diseases (type 2), since they increase venous return to the LA and incur a risk of overload and acute pulmonary edema. PDE5 inhibitors are the only appropriate solution in this situation, provided that the transpulmonary gradient (mPAP – PAWP) is > 12 mmHg.
In cases of extreme right-sided overload, the RV can be decompressed and LV preload improved by atrial septostomy (iatrogenic ASD), though at the cost of arterial desaturation. Its benefit is dependent on the balance struck between O2 delivery improvement on one hand and arterial desaturation due to R-to-L shunting on the other [1].
Ventilation and pulmonary hypertension
Ventilation requires a compromise between active hyperventilation and maintaining low mean intrathoracic pressure (ITP). If the tidal volume is low, there is a risk of atelectasis, hypercapnia and increased PVR in the small perialveolar vessels. If it is high, ITP rises and the large extra-alveolar vessels are compressed due to hyperinflation [15]. Ideally, the tidal volume should be the same as the functional residual capacity (FRC) (Figure 15.68).

Figure 15.68: Trend for pulmonary vascular resistance (PVR) with ventilation. A: Normal situation. At low tidal volume, PVR is raised in the small vessels by hypoventilation and hypercapnia. At high tidal volume, it is raised in the large perialveolar vessels, which are mechanically stretched and compressed. The resultant (red curve) shows that the best compromise is reached at the volume of functional residual capacity (FRC). This represents ventilation at 6-8 mL/kg [15]. B: In subjects with chronic pulmonary hypertension, the artery walls are thickened with increased tunica media musculature, subendothelial proliferation, and fibrosis of the tunica externa (see pulmonary artery cross-section in the inset). The intrathoracic pressure is no longer capable of compressing these vessels. The curve representing the large vessels is therefore flattened and total PVR is no longer U-shaped. It no longer rises at high tidal volume. Therefore, these patients tolerate hyperventilation under IPPV very well.
Adjustments must be made to the tidal volume, rate and ventilation method (pressure-controlled ventilation) to achieve the lowest possible PaCO2 and mean ITP. To that end, the duration of inspirium increases the mean ITP more than the peak inspiratory pressure value [28]. However, the right side of the graph in Figure 15.68 does not genuinely apply to patients with PAH, since the thickened, rigid walls of their pulmonary vessels prevent any compression by high tidal volume ventilation. There is therefore no reason to fear an additional increase in PAP if mechanical hyperventilation is used.
Regional anaesthesia is often offered due to concerns over the negative impact of general anaesthesia on ventilation. In this particular case, such concerns are unfounded. Indeed, the increase in afterload for the RV induced by IPPV is very small compared to its normal afterload – adding mean ITP of 10 mmHg to a mean PAP of 50 mmHg changes haemodynamic conditions less than if the mean PAP is normal (≤ 15 mmHg). Moreover, with IPPV, it is possible to hyperventilate patients and thus reduce their PVR. In contrast, arterial vasodilation prompted by neuraxial anaesthesia would exacerbate the shunt's R-to-L component, causing arterial desaturation, and increasing cyanosis [2]. Patients’ haemodynamic reaction to IPPV may be tested preoperatively by instructing them to perform a Valsalva manoeuvre once the arterial catheter is in place and observing changes in arterial pressure. Usually, respiratory variations are reduced and mean pressure (MAP) is stabilised at a value that is very slightly lower (< 10%) than its value during spontaneous breathing. This is indicative of good tolerability to IPPV.
Anaesthesia and pulmonary hypertension
Anaesthetic agents have little impact on pulmonary circulation, except ketamine, N2O and desflurane, which cause sympathetic stimulation and increased PVR in adults [12,14]. Recommendations for anaesthetising patients with PAH may be set out as follows [11,15,16,27,28]:
© BETTEX D, CHASSOT PG, January 2008, last update May 2018
References
In cases of extreme right-sided overload, the RV can be decompressed and LV preload improved by atrial septostomy (iatrogenic ASD), though at the cost of arterial desaturation. Its benefit is dependent on the balance struck between O2 delivery improvement on one hand and arterial desaturation due to R-to-L shunting on the other [1].
| Pulmonary arterial hypertension (PAH) in congenital heart disease patients |
| Definition of PAH: mean PAP > 25 mmHg at rest. Three main mechanisms: - Passive hypertension secondary to left heart dysfunction - Resistance-based hypertension secondary to an increase in PVR - Hyperkinetic hypertension secondary to increased blood flow (L-to-R shunt) Eisenmenger's syndrome (irreversible PAH): mean PAP > 50 mmHg, PVR > 800 dynes•cm•s-5 and bidirectional shunt with cyanosis (L-to-R shunt + R-to-L component) Inoperability criterion: PVR/SVR ratio > 0.7 Haemodynamic characteristics - Systemic hypotension: exacerbation of the shunt's R-to-L component - Low and fixed pulmonary blood flow, hypoxaemia on exercise or in the event of hypovolaemia - Pulmonary vascular reactivity preserved - RVH: RV output dependent on its preload, intolerance of hypovolaemia - Systemic hypotension: risk of RV ischaemia, ventricular interdependence imbalance |
| Treatment of pulmonary hypertension |
| Correction of acidosis, hypothermia, stress and pain. Ventilation and inhalation (no systemic effect) - Hyperventilation with low intrathoracic pressure - NO• (10-30 ppm) - Nebulised prostacyclin (iloprost), milrinone or nitroglycerin Pulmonary vasodilators (risk of systemic hypotension) - Prostacyclins (epoprostenol, treprostinil) - Phosphodiesterase-5 inhibitors (sildenafil) - Endothelin receptor antagonists (bosentan) - Phosphodiesterase-3 inhibitors (milrinone) - Cardiac stimulants: milrinone + epinephrin, dobutamine, levosimendan - Less effective: nitroglycerin, nesiritide, riociguat, Mg2+ - Calcium channel blockers contraindicated for congenital heart patients Systemic vasoconstrictors - Norepinephrin - Vasopressin Non-drug procedures - ECMO, right ventricular assist device - Atrial septostomy - Pulmonary thromboendarterectomy post-embolism - Lung or heart-lung transplant The key to improving haemodynamics lies not only in lowering PAP, but also in lowering CVP and increasing stroke volume, which indicates improved RV function. |
Ventilation and pulmonary hypertension
Ventilation requires a compromise between active hyperventilation and maintaining low mean intrathoracic pressure (ITP). If the tidal volume is low, there is a risk of atelectasis, hypercapnia and increased PVR in the small perialveolar vessels. If it is high, ITP rises and the large extra-alveolar vessels are compressed due to hyperinflation [15]. Ideally, the tidal volume should be the same as the functional residual capacity (FRC) (Figure 15.68).
Figure 15.68: Trend for pulmonary vascular resistance (PVR) with ventilation. A: Normal situation. At low tidal volume, PVR is raised in the small vessels by hypoventilation and hypercapnia. At high tidal volume, it is raised in the large perialveolar vessels, which are mechanically stretched and compressed. The resultant (red curve) shows that the best compromise is reached at the volume of functional residual capacity (FRC). This represents ventilation at 6-8 mL/kg [15]. B: In subjects with chronic pulmonary hypertension, the artery walls are thickened with increased tunica media musculature, subendothelial proliferation, and fibrosis of the tunica externa (see pulmonary artery cross-section in the inset). The intrathoracic pressure is no longer capable of compressing these vessels. The curve representing the large vessels is therefore flattened and total PVR is no longer U-shaped. It no longer rises at high tidal volume. Therefore, these patients tolerate hyperventilation under IPPV very well.
Adjustments must be made to the tidal volume, rate and ventilation method (pressure-controlled ventilation) to achieve the lowest possible PaCO2 and mean ITP. To that end, the duration of inspirium increases the mean ITP more than the peak inspiratory pressure value [28]. However, the right side of the graph in Figure 15.68 does not genuinely apply to patients with PAH, since the thickened, rigid walls of their pulmonary vessels prevent any compression by high tidal volume ventilation. There is therefore no reason to fear an additional increase in PAP if mechanical hyperventilation is used.
Regional anaesthesia is often offered due to concerns over the negative impact of general anaesthesia on ventilation. In this particular case, such concerns are unfounded. Indeed, the increase in afterload for the RV induced by IPPV is very small compared to its normal afterload – adding mean ITP of 10 mmHg to a mean PAP of 50 mmHg changes haemodynamic conditions less than if the mean PAP is normal (≤ 15 mmHg). Moreover, with IPPV, it is possible to hyperventilate patients and thus reduce their PVR. In contrast, arterial vasodilation prompted by neuraxial anaesthesia would exacerbate the shunt's R-to-L component, causing arterial desaturation, and increasing cyanosis [2]. Patients’ haemodynamic reaction to IPPV may be tested preoperatively by instructing them to perform a Valsalva manoeuvre once the arterial catheter is in place and observing changes in arterial pressure. Usually, respiratory variations are reduced and mean pressure (MAP) is stabilised at a value that is very slightly lower (< 10%) than its value during spontaneous breathing. This is indicative of good tolerability to IPPV.
Anaesthesia and pulmonary hypertension
Anaesthetic agents have little impact on pulmonary circulation, except ketamine, N2O and desflurane, which cause sympathetic stimulation and increased PVR in adults [12,14]. Recommendations for anaesthetising patients with PAH may be set out as follows [11,15,16,27,28]:
- Deep anaesthesia (stress-free) using fentanil and a hypnotic agent (halogenated agent, propofol);
- Hyperventilation with low intrathoracic pressure (IPPV), respiratory alkalosis – use of NO (10-20 ppm);
- Normothermia;
- Maintain preload: avoid any hypovolaemia;
- Maintain SVR (vasoconstrictor): coronary perfusion pressure, reduction of the shunt’s R-to-L component, repositioning of the interventricular septum;
- Monitoring: an arterial line and CVP monitoring are necessary. A system for measuring cardiac output is necessary for major surgery. However, the pulmonary catheter may be difficult or even dangerous to place depending on the patient’s anatomical configuration. A PiCCO-type device is very useful.
- Transfusion threshold: Hb 100 g/L.
| Anaesthesia and PAH |
| Avoid any increase in PVR: hypoventilation, hypercapnia, atelectasis, high intrathoracic pressure, acidosis, hypothermia, sympathetic stimulation, stress, pain Anaesthesia: - Deep anaesthesia and analgesia - Hyperventilation with low intrathoracic pressure, respiratory alkalosis, normoxia, NO - Maintain preload - Maintain SVR - Normothermia - Spinal anaesthesia poorly tolerated, epidural possible if sympathetic blockade is installed very slowly IPPV is well tolerated for 3 reasons: - The increase in right-side afterload is low compared to the chronically high PAP - The hypertrophied RV can withstand increased afterload - Thickened and fibrotic pulmonary vessels are not compressible by IPPV |
© BETTEX D, CHASSOT PG, January 2008, last update May 2018
References
- ALTHOFF TF, KNEBEL P, PANDA A, et al. Long-term follow-up of a fenestrated Amplatzer atrial septal occluder in pulmonary arterial hypertemsion. Chest 2008; 133:283-5
- AMMASH NA, CONNOLLY HM, ABEL MD, WARNES CA. Noncardiac surgery in Eisenmenger syndrome. J Am Coll Cardiol 1999; 33:222-7
- BARR FE, MACRAE D, Inhaled nitric oxide and related therapies. Pediatr Crit Care Med 2010; 11(Suppl): S30-S36
- BAUMGARTNER H, BONHOEFFER P, DE GROOT NMS, et al. ESC Guidelines for the management of grown-up congenital heart disease (new version 2010). Eur Heart J 2010; 31:2915-57
- BELZER DT, KORT HW, DAY RW, et al. Inhaled nitric oxide as a preoperative test (INOP Test I): the INOP Test Study Group. Circulation 2002; 106(Suppl I):S76-81
- BENZA RL, MILLER DP, BARST RJ, et al. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from REVEAL. Chest 2012; 142:448-56
- BLAISE G, LANGLEBEN D, HUBERT B. Pulmonary arterial hypertension. Pathophysiology and anesthetic approach. Anesthesiology 2003; 99:1415-32
- BRONICKI RA, BADEN HP. Pathophysiology of right ventricular failure in pulmonary hypertension. Pediatr Crit Care Med 2010; 11(Suppl):S15-S22
- BUNDTS W, VAN PELT N, GILLYNS H, et al. Residual pulmonary vasoreactivity to inhaled nitric oxide in patients with severe obstructive pulmonary hypertension and Eisenmenger syndrome. Heart 2001; 86:553-9
- CANNESSON M, EARING MG, COLLANGE V, KERSTEN JR. Anesthesia for noncardiac surgery in adults with congenital heart disease. Anesthesiology 2009; 111:432-40
- CHASSOT PG, BETTEX DA. Anesthesia and adult congenital heart disease. J Cardiothorac Vasc Anesth 2006; 20:414-37
- CIOFOLO M, REIZ S. Circulatory effects of volatile anesthetic agents. Minerva Anaesthesiol 1999; 65:232-8
- DALIENTO L, SOMERVILLE J, PRESBITERO P, et al. Eisenmenger syndrome. Factors relating to deterioration and death. Eur Heart J 1998; 19:1845-55
- EBERT TJ, MUZI M. Sympathetic hyperactivity during desflurane anesthesia in healthy volunteers: a comparison with isoflurane. Anesthesiology 1993; 79:444-53
- FISCHER LG, VAN HAKEN H, BÜRKLE H. Management of pulmonary hypertension: Physiological and pharmacological considerations for anesthesiologists. Anesth Analg 2003; 96:1603-1664 FRANK SM, FLEISHER LA, BRESLOW MJ, et al. Perioperative maintenance of normothermia reduces the incidence of morbid cardiac events. A randomized clinical trial. JAMA 1997; 277: 1127-34
- FRANK SM, FLEISHER LA, BRESLOW MJ, et al. Perioperative maintenance of normothermia reduces the incidence of morbid cardiac events. A randomized clinical trial. JAMA 1997; 277: 1127-34
- GALIÉ N, HUMBERT M, VACHIERY JL, et al. 2015 ESC/ERS Guideline for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2016; 37:67-119
- GALLE N, HOEPER MM, HUMBERT M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2009; 30:2493-537
- GHOFRANI HA, WIEDEMAN R, ROSE F, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension. A randomized controlled trial, Lancet 2002; 360:895-900
- GUAZZI M, BORLAUG BA. Pulmonary hypertension due to left heart disease. Circulation 2012; 126:975-90
- HADDAD F, COUTURE P, TOUSIGNANT C, DENAULT AY. The right ventricle in cardiac surgery, a perioperative perspective: I. Anatomy, physiology and assessment. Anesth Analg 2009; 108:407-21
- HADDAD F, DOYLE R, MURPHY DJ. HUNT SA. Right ventricular function in cardiovascular disease, Part II. Pathophysiology, clinical importance and management of right ventricular failure. Circulation 2008; 117:1717-31
- HOEPER MM, BOGAARD HJ, CONDLIFFE R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013; 62:D42-50
- HOPKINS WE, OCHOA LL, RICHARDSON GW, TRULOCK EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. J Heart Lung Transplant 1996; 15:100-5
- HUMBERT M, SITBON O, SIMONEAU G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004; 351:1425-36
- JEON Y, RYU JH, LIM YJ, et al. Comparative hemodynamic effects of vasopressin and norepinephrine after milrinone-induced hypotension in off-pump coronary artery bypass surgical patients. Eur J Cardiothorac Surg 2006; 29:952-6
- KERBAUL F, RONDELET B, COLLART F, et al. Hypertension artérielle pulmonaire en anesthésie-réanimation. Ann Fr Anesth Réanim 2005; 24:528-40
- LOVELL AT. Anaesthetic implications of grown-up congenital heart disease. Br J Anaesth 2004; 93:129-39
- MICHELAKIS E, TYMCHAK W, LIEN D, et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: Comparison with inhaled nitric oxide. Circulation 2002; 105:2398-403
- OECHSLIN E. Eisenmenger's syndrome. In: GATZOULIS MA, WEBB GD, DAUBENEY PEF, Eds. Diagnosis and management of adult congenital heart disease. Edinburgh: Churchill Livingstone 2003, 363-77
- OPOTOWSKY AR. Clinical evaluation and management of pulmonary hypertension in the adult with congenital heart disease. Circulation 2015; 131:200-10
- PALEVSKY HI, SCHLOO BL, PIETRA CC, et al. Primary pulmonary hypertension: Vascular structure, morphometry, and responsiveness to vasodilator agents. Circulation 1989; 80:1207-21
- POLLESELLO P, PARISSIS J, KIVIKKO M, et al. Levosimendan meta-analyses: is there a pattern in the effect on mortality ? Int J Cardiol 2016; 209: 77-83
- RABINOVITCH M, HAWORTH SG, CASTANEDA AR, et al. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation 1978; 58:1007-22
- RICH S, MCLAUGHLIN VV. The effects of chronic prostacyclin therapy on cardiac output and symptoms in primary pulomonary hypertension. J Am Coll Cardiol 1999; 34:1184-7
- SCHROEDER RA, WOOD GL, PLOTKIN JS, et al. Intraoperative use of inhaled PGI(2) for acute pulmonary hypertension and right ventricular failure. Anesth Analg 2000; 91:291-5
- SIMONEAU G, GATZOULIS MA, ADAIA I,, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62:D34-41
- THUNBERG CA, GAITAN BD, GREWAL A, et al. Pulmonary hypertension in patients undergoing cardiac surgery: pathophysiology, perioperative management and outcome. J Cardiothorac Vasc Anesth 2013; 27: 551-72
- WINTERHALTER M, ANTONIOU T, LOUKANOV T. Management of adult patients with perioperative pulmonary hypertension: technical aspects and therapeutic options. Cardiology 2010; 116:3-9
15. Anesthesia for adult congenital heart disease patients
- 15.1 Introduction
- 15.2 Nomenclature and pathophysiology
- 15.3 Approach by pathology
- 15.3.1 Classification
- 15.3.2 Diagnostic methods
- 15.3.3 Anomalous venous returns
- 15.3.4 Atrial septal defects (ASDs)
- 15.3.5 Atrioventricular canal (AVC) defects
- 15.3.6 Ebstein anomaly
- 15.3.7 Ventricular septal defects (VSDs)
- 15.3.8 Ventricular hypoplasia
- 15.3.9 Tetralogy of Fallot
- 15.3.10 Mixed shunt
- 15.3.11 Pulmonary stenosis
- 15.3.12 Anomalies of the LV ejection pathway
- 15.3.13 Transposition of the great arteries (TGA
- 15.3.15 Coarctation of the aorta
- 15.3.14 Congenitally corrected TGA
- 15.3.16 Arterial abnormalities
- 15.4 General considerations for anesthesia
- 15.5 Conclusions