




























Step 1 of 1
Epidémiologie
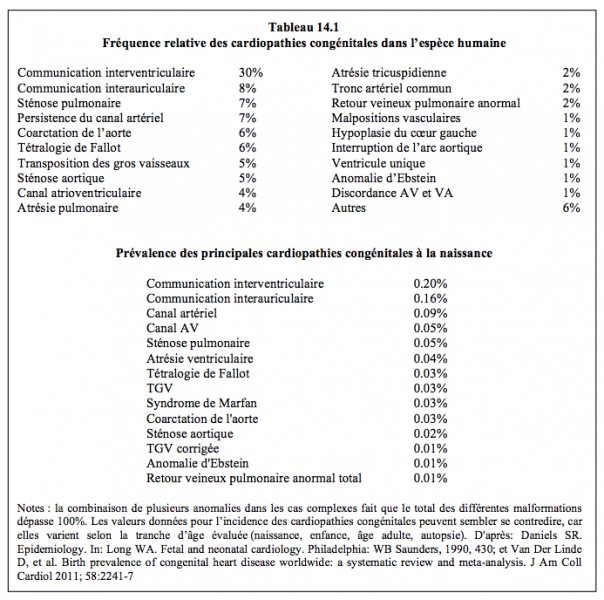
Bien qu'elles paraissent être un sujet fort ésotérique, les cardiopathies congénitales sont assez fréquentes, puisqu’elles surviennent dans 0.5-1% des naissances, si l'on exclut le foramen ovale permérable (incidence 20%) et la bicuspidie aortique (incidence 2%). La prévalence moyenne est de 0.93% en Asie, de 0.82% en Europe, de 0.65% en Amérique du Sud et de 0.2% en Afrique [12]. Les malformations complexes, plus rares, n'apparaissent que chez 1‰ des nouveau-nés [3]. La prévalence chez les enfants nés de mères souffrant elles-mêmes de cardiopathies congénitales est de 3% pour la tétralogie de Fallot, de 6-8% pour la CIV et s’élève jusqu’à 12% pour le canal AV [1,10]. Bien que d’innombrables malformations aient été décrites, les trois quarts d’entre elles font partie des anomalies suivantes: CIV (26%), persistance du canal artériel (14%), CIA (11%), tétralogie de Fallot (9%), sténose pulmonaire (8%), coarctation de l'aorte (6%), transposition des gros vaisseaux (6%) et sténose aortique (5%) [5,12]. La prévalence des principales malformations est indiquée dans le Tableau 14.1. La cardiopathie congénitale doit être considérée comme une affection systémique, car elle est associée dans plus d'un quart des cas à une cascade de malformations au niveau du squelette (dysmorphisme et difficulté d'intubation), du système nerveux central (convulsions), de l'appareil respiratoire (lésions restrictives et obstructives), du foie et des reins (65% des cyanosés souffrent de néphropathie) [2].
Les cardiopathies congénitales relèvent de plusieurs origines et sont associées à plusieurs phénomènes [7,8,11].
- Génétique (décelable dans 30% des cas) : trisomie 21, 13 et 18, syndrome de Noonan et de Williams, microdélétion 22q11, mutations Zic3, FoxH1 ou CFC1, anomalies de la latéralité liées au gène PITX2, erreurs de transcription dans des molécules de différentiation (TGF-β, SMAD), mutations au sein de l'ADN (NKX2.5, MESP1) [7].
- Longue liste de syndromes congénitaux rares fréquemment associés à des malformations cardiaques.
- Maternelle : la rubéole, le diabète, le lupus ou l'alcoolisme chez la mère ; la consommation de thalidomide, de lithium ou de divers anticonvulsivants et anti-épileptiques pendant la grossesse.
- Anomalies de flux : un débit intracardiaque anormal peut influencer le développement des structures d’aval.
- Prématurité.
Mais le plus souvent, on ne retrouve pas de facteur causal directement impliqué. Il s’agit alors d’un défaut de fonctionnement local et aléatoire dans la machinerie complexe de l’embryologie.
La chirurgie cardiaque pédiatrique a fait de tels progrès au cours de ces 30 dernières années que la mortalité opératoire est maintenant globalement inférieure à 5%. La survie jusqu’à l’âge adulte des enfants opérés de cardiopathies congénitales est en moyenne de 80% pour les cardiopathies complexes et de ≥ 98% pour les cas simples [9,11,12,13]. Toutefois, seuls le canal artériel (CA), la communication interauriculaire (CIA) de type ostium secundum et une petite communication interventriculaire (CIV) isolée permettent une correction totale sans séquelle, à la condition d’être opérés avant l’âge de 2 ans (CIV) à 5 ans (CIA) [6]. Pour les autres, l'importance des lésions résiduelles est fonction de l'âge auquel a eu lieu la correction chirurgicale: plus l'enfant est opéré tôt (< 3 ans), moins les séquelles sont importantes. Pour cette raison, on procède autant que possible à une correction totale dans la petite enfance plutôt qu’à des interventions palliatives. Malgré les risques opératoires très élevés, cette tendance offre aux enfants une qualité de vie quasi-normale, bien que 27% d'entre eux doivent subir une deuxième intervention [4,6]. La transposition des gros vaisseaux (TGV) en est un bon exemple: l'opération de switch aorto-pulmonaire réalisée pendant les trois premières semaines de vie permet un développement ultérieur adéquat (survie à 20 ans ≥ 95%), ce que les interventions de croisement auriculaire (opération de Mustard ou de Senning) réalisées 2 ou 3 ans plus tard ne peuvent pas garantir (survie à 20 ans 56-70%) [6]. Les avancées de la cardio-chirurgie pédiatrique sont bien illustrés par la survie à 16 ans des enfants opérés de truncus arteriosus ou d'hypoplasie de l'arc aortique: de 20% dans les années '70, elle est passée à 95% depuis les années 2000 [6]. Malheureusement, il existe aussi un certain nombre de pathologies pour lesquelles s’imposent des opérations palliatives, souvent itératives, telle l’hypoplasie du coeur gauche ou du cœur droit; les interventions en plusieurs stades (Norwood, Fontan) permettent de prolonger la vie, mais n’assurent pas un avenir adulte normal (survie à 20 ans 80%) [6]. Seule la transplantation assure une correction complète.
La prise en charge des enfants souffrant de malformation cardiaque réclame des équipes spécialisées et des connaissances particulières qui sont différentes de celles que l’on acquière dans le domaine conventionnel de la cardiologie, de l’anesthésie et de la pédiatrie. Néanmoins, même s'il ne pratique pas dans un centre spécialisé, chaque anesthésiste peut rencontrer un de ces malades à l'occasion d'une opération non-cardiaque urgente telle une appendicectomie ou une réduction de fracture.
Ce chapitre insiste volontairement sur les aspects cardiologiques et chirurgicaux, car seule une bonne compréhension de la cardiopathie et de sa correction chirurgicale permet à l’anesthésiste de gérer adéquatement la prise en charge de l’enfant au cours de l’intervention. Les malformations cardiaques sont un domaine où aucune recette passe-partout n’est applicable pour l’anesthésie. Les Chapitres 14 et 15 traitant des mêmes pathologies mais à des âges différents, il est inévitable qu'ils se recoupent largement et qu'ils contiennent des répétitions de l'un à l'autre.
| Cardiopathies congénitales de l’enfant |
| La prévalence des cardiopathies congénitales est de 0.8% des naissances (1‰ pour les cas complexes). Actuellement, la chirurgie offre une survie jusqu’à l’âge adulte de 90% pour les cardiopathies complexes et de ≥ 98% pour les cas simples. Une cardiopathie opérée est une cardiopathie corrigée, mais non guérie. Seuls le canal artériel, la CIA et la CIV simples opérés avant l’âge de 3 ans permettent un avenir sans séquelle. |
© BETTEX D, BOEGLI Y, CHASSOT PG, Juin 2008, dernière mise à jour Mai 2018
Références
- BAUMGARTNER H, BONHOEFFER P, DE GROOT NMS, et al. ESC Guidelines for the management of grown-up congenital heart disease (new version 2010). Eur Heart J 2010; 31:2915-57
- BHATT AB, FOSTER E, KUEHL K, et al. Congenital hesart disease in older adult. A Scientific Statement from the American Heart Association. Circulation 2015; 131:1884-931
- BRICKNER EM, HILLIS LD, LANGE RA. Congenital heart disease in adults. First of two parts. N Engl J Med 2000; 342:1-12
- CASTANEDA AR, JONAS RA, MAYER JE, HANLEY FL. Cardiac Surgery of the Neonate and Infant. Philadelphia:WB Saunders, 1994, 23
- DANIELS SR. Epidemiology. In: LONG WA. Fetal and neonatal cardiology. Philadelphia: WB Saunders, 1990, 430
- ERIKSSEN G, LIESTOL K, SEEM E, et al. Achievements in congenital heart defect surgery: a prospective, 40-year study of 7'038 patients. Circulation 2015; 131:337-46
- KLOESEL B, DINARDO JA, BODY SC. Cardiac embryology and molecular mechanisms of congenital heart disease – A primer for anesthesiologists. Anesth Analg 2016; 123:551-69
- KUSSMAN BD, HOLZMAN RS. Cardiac embryology : Understanding congenital heart disease for the noncardiac anesthesiologist. Sem Cardiothorac Vasc Anesth 2001 ; 5 : 2-20
- MARELLI AJ, MACKIE AS, IONESCU-ITTU R, et al . Congenital heart disease in the general population: changing prevalence and age distribution. Circulation 2007; 115:163-72
- SILVERSIDES CK, DORE A, POIRIER N, et al. Canadian Cardiovascular Society 2009 Consensus Conference on the management of adults with congenital heart disease: Shunt lesions. Can J Cardiol 2010; 26:e70-e79
- TRIEDMAN JK, NEWBURGER JW. Trends in congenital heart disease. The next decade. Circulation 2016; 133:2716-33
- VAN DER LINDE D, KONINGS EE, WITZENBURG M, et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol 2011; 58:2241-7
- WARNES CA, LIBERTHSON R, DANIELSON GK, et al: Task Force 1: The changing profile of congenital heart disease in adult life. J Am Coll Cardiol 2001; 37:1170-5
14. Anesthésie pour la chirurgie cardiaque pédiatrique
- 14.1 Introduction
- 14.2 Physiopathologie
- 14.3 Stratégies hémodynamiques
- 14.3.1 Classification
- 14.3.2 Shunt G → D et débit pulmonaire élevé
- 14.3.3 Hypertension pulmonaire pédiatrique
- 14.3.4 Shunt droit → gauche et débit pulmonaire abaissé
- 14.3.5 Shunt droit → gauche cyanogène et débit systémique abaissé
- 14.3.6 Shunt cyanogène mixte
- 14.3.7 Cardiopathies sans shunt : obstructions et valvulopathies
- 14.3.8 Options thérapeutiques chez le nouveau-né
- 14.3.9 Pharmacothérapie
- 14.4 Techniques d'anesthésie
- 14.5 La CEC chez l'enfant
- 14.6 Approche par pathologie
- 14.6.1 Introduction
- 14.6.2 Repères anatomiques
- 14.6.3 Retours veineux anormaux
- 14.6.4 Communication interauriculaire (CIA)
- 14.6.5 Canal atrio-ventriculaire (CAV)
- 14.6.6 Maladie d'Ebstein
- 14.6.7 Anomalies des valves auriculo-ventriculaires
- 14.6.8 Communication interventriculaire (CIV)
- 14.6.9 Hypoplasie ventriculaire
- 14.6.10 Tétralogie de Fallot
- 14.6.11 Ventricule droit à double issue
- 14.6.12 Atrésie pulmonaire
- 14.6.13 Anomalies de la voie éjectionnelle gauche
- 14.6.14 Transposition des gros vaisseaux
- 14.6.15 Truncus arteriosus
- 14.6.16 Coarctation de l'aorte
- 14.6.17 Anomalies artérielles
- 14.6.18 Transplantation cardiaque
- 14.7 Conclusions