




























Step 3 of 3
Stimulation neuro-humorale
Dans l'insuffisance systolique, la baisse de l'éjection se manifeste par la fatigabilité. Elle provoque une tachycardie compensatrice, une vasoconstriction périphérique et une rétention de sodium, puis une hypoperfusion des organes engendrant la cyanose, l'oligurie et la confusion mentale [17]. L'insuffisance diastolique, qui peut survenir sans perturbation de la fonction systolique, se traduit par une pression diastolique ventriculaire élevée et une stase en amont. Elle se caractérise cliniquement par une dyspnée [2,11,20]. L'insuffisance cardiaque congestive (ICC), cliniquement caractérisée par les oedèmes et la dyspnée, est donc une symptomatologie commune à la dysfonction ventriculaire systolique et diastolique.
Les barorécepteurs de la crosse aortique et des sinus carotidiens règlent le volume sanguin effectif conjointement aux mécanorécepteurs situés dans les cavités cardiaques et l'arbre pulmonaire. Les premiers sont sensibles à la hausse de pression intravasculaire, les seconds à sa baisse. Lorsque la capacité ventriculaire à pomper le sang s'effondre, les premiers sont inhibés et les seconds sont activés. Il en résulte une prédominance du tonus sympathique qui conduit à une tachycardie, à une augmentation de contractilité et à une vasoconstriction artériolaire [12]. Les chémorécepteurs sont également activés: la sensibilité élevée à l'hypoxie et à l'hypercarbie augmente encore la stimulation neuro-humorale.
Stimulation catécholaminergique
L'hypotension et la baisse de la pression pulsée (PAsyst – PAdiast) sont à l'origine d'une stimulation des barorécepteurs et du système sympathique central. Les taux de noradrénaline circulante sont élevés, et sont un marqueur pronostique de l'évolution de la maladie [14]. La stimulation du système catécholaminergique (adrénaline surrénalienne et nor-adrénaline neuronale) et du système rénine-angiotensine-aldostérone est utilisée au maximum afin d'augmenter la [Ca2+]i, et de maintenir la précharge et la pression de perfusion, mais ses effets aggravent l'insuffisance ventriculaire gauche (voir Figure 5.8) [9].
- Surcharge en [Ca2+]i et défaut de recapture par le réticulum sarcoplasmique (manque de SERCA et excès de phospholamban);
- Effet α : augmentation de postcharge;
- Effet β: augmentation de la contractilité et de la mVO2, tachycardie sinusale avec diminution du temps de remplissage diastolique, arythmies;
- A long terme : excès de peroxydes, hypertrophie et dilatation du ventricule, fibrose intersticielle, apoptose.
L'excès de catécholamines plasmatiques conduit par rétro-action à une diminution de l'activité des récepteurs β1 myocardiques (désensibilisation ou downregulation), dont le nombre diminue de moitié dans l’insuffisance ventriculaire (down-regulation), et ne représente plus que 40% du total des récepteurs (normal : 70-80%). En compensation, la proportion des récepteurs β2 chronotropes positifs et faiblement inotropes positifs augmente à 40% (normal 10-20%) et celle des récepteurs α1 (inotropes positifs) à 20% (normal 10%) [1,4,5,12]. Les récepteurs α2 myocardiques ne sont pas concernés par cette dysrégulation; ils contribuent à la mobilisation du Ca2+ intracellulaire indépendamment de l'AMPc [6,24]. Les taux circulants de nor-adrénaline sont élevés à cause d’une sécrétion présynaptique accrue et d’une recapture diminuée, mais la stimulation sympathique chronique finit par épuiser les réserves myocardiques de la substance [9]. Enfin, les récepteurs au VIP (vasoactive intestinal peptide) sont plus sensibles que la norme et maintiennent la contractilité malgré la baisse de sensibilité des récepteurs β1, mais au prix d'un excès de vasoconstriction [18,19]. Ces données ont des impacts majeurs en clinique [16,22,23].
- Les β-bloqueurs améliorent le pronostic de l'insuffisance cardiaque (frein à la désensibilisation).
- La réponse aux amines β1 est altérée ; la désensibilisation β1 survient après quelques jours d’une perfusion de catécholamines β1 ; l'adrénaline, qui stimule les récepteurs β1, β2 et α1, est en général la seule substance efficace pour le traitement aigu de la décompensation d'une insuffisance cardiaque chronique.
- La déplétion chronique du myocarde en nor-adrénaline diminue l'efficacité des amines indirectes telles la dopamine ou l'éphédrine.
- Lorsqu'une décompensation cardiaque survient chez un malade β-bloqué, la réponse cardiaque à la dopamine est obscurcie, laissant apparente la stimulation α, d'où prédominance de l'effet vasoconstricteur périphérique.
- Les inhibiteurs des phosphodiestérases-3 (amrinone, milrinone) et le levosimendan (sensibilisateur calcique) sont efficaces lors d’insuffisance ventriculaire ou de β-blocage parce qu'ils agissent par une voie indépendante des récepteurs catécholaminergiques.
Stimulation neuro-humorale
L'hypoperfusion rénale et la stimulation sympathique conduisent à accroître le taux de rénine par vasoconstriction de l'artériole afférente, et déclenchent une vasoconstriction puissante par l'angiotensine II [9]. Celle-ci contribue à la sécrétion d'aldostérone, qui provoque une rétention d'eau et de sodium avec oedèmes périphériques et qui est aussi un puissant inducteur de l’HVG. Cet excès de volume extra- et intracellulaire crée une hyponatrémie qui est mal interprétée par l'hypophyse, laquelle augmente la sécrétion d'ADH: cette élévation du système arginine-vasopressine retient encore davantage d'eau et accroît le degré de vasoconstriction. Le résultat global est une hypervolémie et une vasoconstriction massive (Figure 5.108). Mais ces substances ont également un effet inotrope positif par l'intermédiaire de la sécrétion d'endothéline qu'elles induisent dans l'endocarde [7]; toutefois, ceci n'est pas que bénéfice, car l'endothéline est un vasoconstricteur très puissant et un facteur important dans la genèse de l’HVG [21]. Il existe aussi une diminution de la réponse vasodilatatrice au NO• [15].

Figure 5.108 : Représentation schématique des modifications neuro-humorales dans l’insuffisance systolique du VG (IVsyst) (Forward failure). Les systèmes physiologiques sont de bons moyens de compensation en temps normal, mais établissent des rétro-actions néfastes (encadrées en rouge) dans l’insuffisance gauche chronique. En jaune figurent les points d’action des principales classes de médicaments utilisés dans l’insuffisance cardiaque gauche. Anti-ET : anti-endothéline (bosantan). IEC : inhibiteur de l’enzyme de conversion (captopril, enalapril, etc). ARA : antagoniste du récepteur de l’angiotensine II (sartan).
On voit que des mécanismes compensatoires deviennent eux-mêmes des facteurs aggravants lorsqu’ils dépassent un certain seuil [3]. Il existe toute une série de points d’impact sur lesquels agissent différentes classes de médicaments. Le point d’impact des principaux d’entre eux est mentionné dans la Figure 5.108. Les mêmes phénomènes rétroactifs existent lors d’insuffisance ventriculaire diastolique (Figure 5.109).
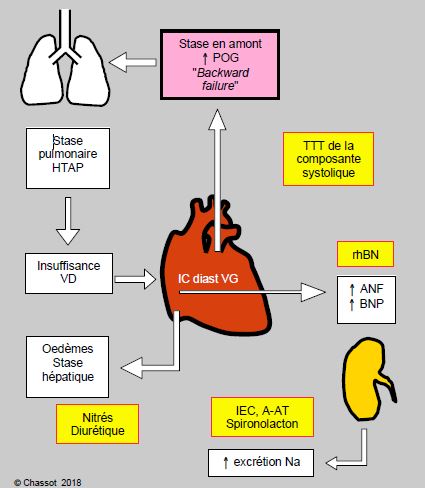
Figure 5.109 : Représentation schématique des modifications neuro-humorales dans l’insuffisance diastolique du VG (Backward failure) avec les points d’action des principales classes de médicaments utilisés dans l’insuffisance cardiaque gauche. rhBNP : recombinant human Brain Natriuretic Peptide (neseritide). IEC : inhibiteur de l’enzyme de conversion (captopril, enalapril, etc). A-AT : inhibiteur du récepteur de l’angiotensine II (losartan). HTAP : hypertension artérielle pulmonaire.
La vasoconstriction systémique déclenchée par l'angiotensine II, la vasopressine et l'endothéline tend à maintenir un flux préférentiel vers le cerveau, le cœur et les poumons au détriment des viscères, des reins, des muscles et de la peau. Les peptides natriurétiques auriculaires (ANF, atrial natriuretic factor) sont des contre-régulateurs de la stimulation sympathique neuro-humorale. Ils sont stimulés par la distension auriculaire et induisent une vasodilatation artérielle et une excrétion rénale de sodium et d'eau. Ces peptides sont détruits par la néprilysine, une peptidase membranaire également active contre l'angiotensine II. Cette action est à la base d'un nouveau médicament, le sacubitril, qui inhibe la dégradation des peptides natriurétiques et qui, en association au valsartan, est une nouvelle piste dans le traitement de l'insuffisance cardiaque par vasodilatation artériolaire [12].
Facteurs natriurétiques et vasodilatateurs
Trois substances sécrétées par la stimulation sympathique modulent les effets hypertenseurs.
- Les facteurs natriurétiques auriculaires, dont la sécrétion est stimulée par la distension des cavités cardiaques, et non par la pression auriculaire [10] : le facteur natriurétique auriculaire (ANF: atrial natriuretic factor) et le facteur natriurétique cérébral (BNP: brain natriuretic peptide). Ils assurent une fuite sodique et diminuent le volume circulant. Ils ont 4 effets majeurs :
- Augmentation de la perfusion rénale et diurèse;
- Baisse des résistances artérielles;
- Effet bloqueur de l’endothéline;
- Inhibition de la sécrétion d’aldostérone.
- Les prostaglandines ; elles ont un effet vasodilatateur systémique et pulmonaire ; leur sécrétion est bloquée par les AINS.
- L'angiotensine 1-7 (AT1-7), issue de l'AT-II, qui a des effets inverses de celle-ci: vasodilatation, production de NO, effet anti-inflammatoire, inhibition de la croissance et de la fibrose [8].
D'autre part, l'augmentation du taux de 2,3-DPG déplace la courbe de dissociation de l'hémoglobine vers la droite: l'affinité de l'O2 pour l'Hb est diminuée, donc sa distribution en périphérie est facilitée. Le BNP est libéré dans la circulation lors de défaillance ventriculaire. Un taux supérieur à 200 pg/ml est habituellement considéré comme significatif pour le diagnostic d’insuffisance cardiaque [13].
| Stimulation neuro-humorale |
| L'insuffisance hémodynamique engendrée par la dysfonction ventriculaire stimule le système sympathique, le système rénine-angiotensine et la sécrétion de catécholamines avec pour effet: tachycardie, vasoconstriction (augmentation de postcharge), rétention de sodium et d'eau (hypervolémie et hyponatrémie), désensibilisation des récepteurs β1, augmentation du taux de récepteurs β2 et α1 (les amines à effet β1 pur perdent leur efficacité). |
© CHASSOT PG Août 2010, dernière mise à jour Novembre 2018
Références
- BOHM M. Catecholamines refractoriness and their mechanisms in cardiocirculatory shock and chronic heart failure. J Thorac Cardiovasc Surg 1998; 46(suppl 2):270-5
- BONOW RO, UDELSON JE. Left ventricular diastolic dysfunction as a cause of congestive heart failure. Mechanisms and management. Ann Intern Med 1992; 117:502-10
- BRAUNWALD E. Heart failure. J Am Coll Cardiol HF 2013; 1:1-20
- BRISTOW MR. Antiadrenergic therapy of chronic heart failure. Surprises and new opportunities. Circulation 2003; 107:1100-2
- BRISTOW MR, GINSBURG R, MINOBE W, et al. Decreased catecholamine sensitivity and beta-adrenergic receptor density in failing human hearts. N Engl J Med 1982; 307:205-11
- BRUCKNER R, MUGGE A, SCHOLZ H. Existence and functional role of alpha1-adrenoreceptors in the mammalian heart. J Mol Cell Cardiol 1985; 17:639-45
- DE HERT SG, GILLEBERT TC, ANDRIES LG, BRUTSAERT DL. Role of the endocardial endothelium in the regulation of myocardial function. Anesthesiology 1993; 79:1354-66
- FARAG E, MAHESHWARI K, MORGAN J, et al. An update of the role of rennin angiotensin in cardiovascular homeostasis. Anesth Analg 2015; 120:275-92
- FLORAS JS, PONIKOWSKI P. The sympathetic/parasympathetic imbalance in heart failure with reduced ejection fraction. Eur Heart J 2015; 36:1974-82
- GOTTLIEB S, KUKIN ML, AHERN D. Prognostic importance of atrial natriuretic peptide in patients with chronic heart failure. J Am Clin Cardiol 1989; 10:437-44
- GROSSMAN W. Diastolic dysfunction in congestive heart failure. N Engl J Med 1991; 325:1557-64
- HARTUPEE J, MANN DL. Neurohumoral activation in heart failure with reduced ejection fraction. Nat Rev Cardiol 2017; 14:30-8
- HUTFLESS R, KASANEGRA R, MADANI M, et al. Utility of B-type natriuretic peptide in predicting postoperative complications and outcomes in patients undergoing heart surgery. J Am Coll Cardiol 2004; 43:1873-9
- KAYE DM, LEFKOVITZ J, JENNINGS GL, et al. Adverse consequences of high sympathetic nervous activity in the failing human heart. J Am Coll Cardiol 1995; 26:1257-63
- KUBO S.H., RECTOR T.S., BANK A.J. – Endothelium-dependent vasodilatation is attenuated in patients with heart failure. Circulation 1991; 84:1589-92
- LOWES BD, TSEVTKOVA T, EICHHORN EJ, et al. Milrinone versus dobutamine in heart failure subjects treated chronically with carvedilol. Int J Cardiol 2001; 81:141-9
- McMURRAY JJV. Systolic heart failure. N Engl J Med 2010; 362:228-38
- O’ROURKE MF, SAFAR ME. Relationship between aortic stiffening and microvascular disease in brain and kidney. Cause and logic of therapy. Hypertension 2005; 46:200-4
- O'ROURKE MF, SEWARD JB. Central arterial pressure and arterial pressure pulse: new views entering the second century after Korotkov. Mayo Clin Proc 2006; 81:1057-48
- RAHIMTOOLA SH. The hibernating myocardium. Am Heart J 1989; 117:211-21
- SAKAI S, MIYAUCHI T, KOBAYASHI M, et al. Inhibition of myocardial endothelin pathway improves long-term survival in heart failure. Nature 1996; 384:353-5
- TOLLER WG, STRANZ C. Levosimendan, a new inotropic and vasodilator agent. Anesthesiology 2006; 104:556-69
- TOLLER W, HERINGLAKE M, GUARRACINO F, et al. Preoperative and perioperative use of levosimendan in cardiac surgery: European expert opinion. Int J Cardiol 2015; 184:323-36
- VAGO T, BEVILACQUA M, NORBIATOR G. Identification of an adrenergic receptors on sarcolemma from normal subjects and patients with idiopathic dilated cardiomyopathy. Circ Res 1989; 64:474-81
05 Physiopathologie cardio-vasculaire
- 5.1 Préambule
- 5.2 Couplage de l'excitation et de la contraction myocardiques
- 5.3 La contraction myocardique
- 5.4 Mécanique ventriculaire
- 5.5 Physiopathologie de la systole
- 5.6 Physiopathologie de la diastole
- 5.7 Remplissage veineux
- 5.8 Interactions cardio-respiratoires
- 5.9 Dysfonction ventriculaire gauche
- 5.10 Fonction ventriculaire droite et circulation pulmonaire
- 5.11 Perfusion coronarienne et ischémie myocardique
- 5.12 Conclusions