




























Step 5 of 6
Hypertension pulmonaire
Les conditions de la circulation pulmonaire sont significativement modifiées par l’augmentation chronique de la pression auriculaire gauche au cours de l’évolution des valvulopathies mitrales et aortiques [1].
- L’hypertension veineuse pulmonaire passive et l’augmentation chronique de la pression capillaire (Pcap) jusqu’à une valeur de 18-20 mmHg conduisent à une stase extravasculaire interstitielle avec altération des échanges gazeux et dyspnée. L’œdème intertitiel pulmonaire apparaît dès que la Pcap dépasse 20 mmHg, et l’œdème alvéolaire au-delà de 25 mmHg.
- La pression veineuse pulmonaire chroniquement maintenue au-dessus de 20 mmHg (hypertension pulmonaire postcapillaire) induit une augmentation réactionnelle des résistances artérielles pulmonaires (RAP) de nature myogénique et neurovégétative. Avec le développement progressif d’une fibroélastose intimale, il s’installe une hypertension artérielle pulmonaire (HTAP) de type précapillaire qui est hors de proportion avec l’hypertension veineuse. Cette HTAP précapillaire est probablement adaptative : elle diminue le flux sanguin capillaire et limite le risque d’œdème pulmonaire.
- Des lésions chroniques de fibrose interstitielle pulmonaire abaissent la compliance pulmonaire et augmentent le travail respiratoire, qui représente jusqu'à 25 % de la VO2 totale. Le volume courant et la capacité vitale diminuent. Les rapports V/Q sont altérés et la PaO2 baisse comme dans une pathologie de type restrictif. L’hypoxie et l’hypercapnie ayant un effet vasoconstricteur artériel pulmonaire, le système s’installe dans un cercle vicieux.
L’HVG et le défaut de compliance ventriculaire élèvent la pression dans l’OG, qui se dilate progressivement. Normalement, la contraction auriculaire élève momentanément la pression télédiastolique dans le VG pour mettre sous une tension optimale l’épaisse paroi ventriculaire sans que la pression moyenne de l’oreillette n’atteigne des valeurs qui seraient responsables d’une hypertension veineuse pulmonaire. En l’absence de rythme sinusal, la pression auriculaire moyenne nécessaire à remplir le VG est bien plus élevée, et l’effet protecteur sur la circulation pulmonaire est perdu.
L'hypertension pulmonaire (PAPmoy > 25 mmHg au repos, RAP > 240 dynes•s•cm-5) augmente la postcharge du VD [3]. En l’absence d’hypertrophie compensatrice, la pression systolique maximale que peut développer un VD normal est 50 mmHg. L’HTAP entraîne une cascade de conséquences : hypertrophie droite (HVD) et dilatation du VD, insuffisance tricuspidienne, dilatation de l’OD et stase veineuse systémique. Lorsqu’il s’hypertrophie, le VD ressemble de plus en plus au VG, et son débit devient dépendant de sa précharge. A son tour, la surcharge du VD retentit sur le remplissage du coeur gauche par plusieurs phénomènes : 1) le retour veineux à l’OG est diminué parce que le débit pulmonaire est limité, 2) le septum interventriculaire bombe dans la cavité gauche et limite son expansion diastolique (effet Bernheim), 3) l’insuffisance tricuspidienne due à la dilatation droite fait bomber le septum interauriculaire dans l’OG. La perfusion coronarienne du VD, qui est systolo-diastolique, est compromise lorsque sa pression intracavitaire systolique se rapproche de la pression moyenne aortique ; en cas de défaillance, elle doit être maintenue par une perfusion de vasoconstricteurs (nor-adrénaline), ou par une contre-pulsion intra-aortique [2].
Même si l'hypertension pulmonaire est fixée, sa composante vasospastique réactionnelle est importante et réagit aux mesures vasodilatatrices pulmonaires habituelles : hyperventilation, alcalose, diminution du stress sympathique et vasodilatateurs pulmonaires. Le traitement de l’hypertension pulmonaire est décrit dans le Tableau 12.11 (voir Chapitre 12, Hypertension pulmonaire, traitement). Au cours de la CEC, toute une série de substances vasoactives sont libérées dans la circulation: TNF, interleukines, complément C3a, etc. Elles provoquent une vasodilatation systémique et une vasoconstriction pulmonaire qui apparaissent au moment de la mise en charge du coeur, et qui sont facilement aggravées par l’administration de protamine. La plastie et le remplacement de la valve aortique ou de la valve mitrale abaissent immédiatement la pression veineuse pulmonaire (hypertension postcapillaire), mais ne corrigent pas l’HTAP artérielle précapillaire, qui reste élevée pendant plusieurs semaines. La fibroélastose intimale persiste à long terme.
Le pronostic clinique et le risque de défaillance hémodynamique peropératoire dépendent davantage de la fonction du VD que de la valeur absolue de la PAP (voir Figure 12.7B). En effet, une PAP élevée signifie que le VD est capable de la générer, alors qu’une PAP modérée traduit des RAP modestement élevées ou une dysfonction du VD. Une poussée hypertensive pulmonaire peut conduire à une décompensation ventriculaire droite.
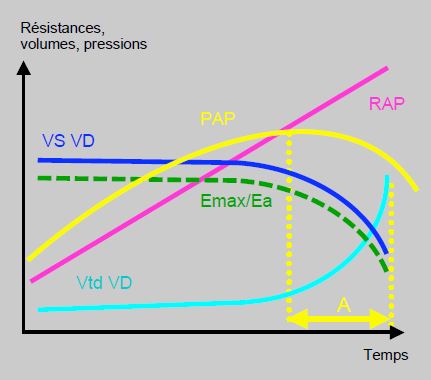
Figure 12.7B : Couplage entre le VD et la circulation pulmonaire. Schématisation de la relation entre les résistances artérielles pulmonaires (RAP), le volume systolique du VD (VS VD), le volume ventriculaire droit (VtdVD), la pression artérielle pulmonaire (PAP) et le rapport Emax/Ea. L’HTAP est fonction de la capacité du VD à générer chroniquement des pressions pulmonaires élevées. Dès que le VD défaille, la PAP tend à redescendre, alors que la situation hémodynamique empire et que les RAS continuent à augmenter. Dans le laps de temps A (en jaune), la PAP mesurée est plus basse que précédemment, non par amélioration de la situation mais pas baisse de fonction du VD [5].
Pour la prise en charge clinique, il importe de distinguer trois situations différentes qui peuvent conduire toutes trois à une défaillance du ventricule droit.
- Infarctus ou défaillance primaire du VD : la priorité est au maintien d’une pression de perfusion coronarienne suffisante (vasoconstricteur systémique) et d’une optimalisation de la précharge (PVC 10-15 mmHg) pour obtenir le meilleur débit. La contractilité est améliorée par des inodilatateurs (dobutamine, milrinone, levosimendan). L’infarctus droit est plus fréquent en cas d’HVD [4].
- Excès de postcharge : défaillance aiguë sur embolie pulmonaire, par exemple, entraînant une insuffisance congestive du VD. La priorité est de baisser la précharge droite (nitroglycérine, position en contre-Trendelenburg), de soutenir l’hémodynamique droite (dobutamine, milrinone ± adrénaline, nor-adrénaline, levosimendan) et d’éviter une aggravation des RAP.
- Poussée d’hypertension pulmonaire en cas d’HTAP chronique avec hypertrophie ventriculaire droite. La priorité est de baisser les RAP; la précharge doit rester élevée car le VD hypertrophié fonctionne sur une courbe de Starling analogue à celle du VG. Contrairement aux deux situations précédentes, la ventilation en pression positive est ici bien tolérée, la postcharge du VD étant déjà chroniquement élevée.
Le traitement spécifique de la décompensation droite est mentionné dans le Tableau 12.6.

| Hypertension artérielle pulmonaire (HTAP) |
|
La stase gauche sur valvulopathie mitrale ou aortique entraîne une élévation chronique de la POG et de la pression veineuse pulmonaire (HTAP postcapillaire) avec œdème interstitiel et éventuellement alvéolaire. L'HTAP veineuse conduit à une augmentation réflexe des RAP et à une HTAP précapillaire.
L'HTAP (PAPmoy > 25 mmHg au repos, RAP > 240 dynes•s•cm-5) provoque une dilatation et une hypertrophie du VD (HVD). Plus il s'hypertrophie, plus le VD fonctionne de manière analogue au VG (débit dépendant de la précharge, résistance à la postcharge).
Le pronostic clinique dépend davantage de la fonction du VD que de la valeur absolue de la PAP.
|
© CHASSOT PG, BETTEX D, Août 2011, dernière mise à jour Août 2018
Références
- GUAZZI M, BORLAUG BA. Pulmonary hypertension due to left heart disease. Circulation 2012; 126:975-90
- HADDAD F, DOYLE R, MURPHY DJ. HUNT SA. Right ventricular function in cardiovascular disease, Part II. Pathophysiology, clinical importance and management of right ventricular failure. Circulation 2008; 117:1717-31
- HOEPER MM, BOGAARD HJ, CONDLIFFE R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013; 62:D42-50
- KINCH JW, RYAN TJ. Right ventricular infarction. N Engl J Med 1994; 330:1211-7
- VONK NOORDERGRAAF A, et al. The relationship between the right ventricle and its load in pulmonary hypertension. J Am Coll Cardiol 2017; 69:236-43
11. Anesthésie et valvulopathies
- 11.1 Introduction : prévalence et risques des valvulopathies
- 11.2 Rappel physiopathologique général
- 11.3 Imagerie valvulaire
- 11.4 Situations particulières
- 11.5 Chirurgie valvulaire
- 11.6 Insuffisance mitrale
- 11.6.1 Etiologie de l'insuffisance mitrale
- 11.6.2 Physiopathologie
- 11.6.3 Manifestations cliniques
- 11.6.4 Echocardiographie de l'insuffisance mitrale
- 11.6.5 Indications et résultats opératoires
- 11.6.6 Principes pour l'anesthésie
- 11.6.7 CEC et post CEC
- 11.6.8 IM primaire sur maladie de Barlow
- 11.6.9 IM secondaire sur ischémie myocardique
- 11.6.10 IM secondaire sur défaillance du VG
- 11.7 Sténose mitrale
- 11.8 Sténose aortique
- 11.8.1 Nosologie
- 11.8.2 Physiopathologie
- 11.8.3 Manifestations cliniques
- 11.8.4 Echocardiographie de la sténose aortique
- 11.8.5 Indications et résultats opératoires
- 11.8.6 Principes pour l'anesthésie en chirurgie cardiaque
- 11.8.7 CEC et post-CEC
- 11.8.8 Sténose sous-aortique dynamique
- 11.8.9 Anesthésie pour la chirurgie non-cardiaque
- 11.9 Insuffisance aortique
- 11.10 Maladie aortique
- 11.11 Pathologie tricuspidienne
- 11.12 Pathologie de la valve pulmonaire
- 11.13 Polyvalvulopathies
- 11.14 Conclusions