




























Step 1 of 3
Physiopathologie
En transposant la loi d’Ohm à l’hémodynamique, on obtient l’équation utilisée pour calculer les résistances artérielles : RAP = (PAPm – POG) / DC. Si l’on extrait la pression artérielle pulmonaire de cette équation, on trouve : PAP ≈ POG + (DC • RAP). La pression de l’AP dépend donc de trois facteurs : la pression de l’OG (stase gauche, HTP postcapillaire), le débit pulmonaire (shunt gauche-droit) et les résistances artérielles pulmonaires (hypertension artérielle pulmonaire, HTAP précapillaire).
Classification et pronostic
L'hypertension pulmonaire (HTP), dont la physiopathologie est traitée plus en détail au Chapitre 5 (Hypertension pulmonaire), est définie par une pression moyenne (PAPm) supérieure à 25 mmHg dans l’AP au repos, et des RAP > 240 dynes•s•cm-5 ou > 3 U Wood (valeur normale : 60-120 dynes•s•cm-5, < 2 U Wood) [12,19,30,36]. La PAPO normale, représentant la POG, est < 15 mmHg. Avec l’âge, la PAP s’élève de 1 mmHg par tranche de 10 ans. Elle augmente aussi parallèlement au BMI. La limite de la PAPm normale à 25 mmHg est actuellement remise en question, parce qu'il semble qu'une PAPm de 20-25 mmHg soit déjà liée à une péjoration du pronostic clinique à long terme [29]. La limite supérieure de la PAPm à l'effort est de 30 mmHg pour un débit cardiaque de 10 L/min et des RAP de 3 unités Wood [15].
L’étiologie de l’HTP relève de 5 groupes de causes, selon la classification de l’OMS (Tableau 12.10A) [19,36].

- 1 - Hypertension artérielle pulmonaire (HTAP) proprement dite :
- HTAP primaire (idiopathique familiale), la prévalence de l’HTAP primaire est de 15 cas (France) à 25 cas (Suisse) par million d’habitants (5% des cas d’HTP) [41].
- HTAP des cardiopathies congénitales (10% des cas adultes) ; la fermeture des shunts cardiaques (gauche-droite) est contre-indiquée lorsque le rapport pression systémique/pression pulmonaire est > 0.7 et/ou les RAP > 4.5 U Wood (> 360 dynes•s•cm-5).
- HTAP secondaire des médicaments (anorexigènes comme l’aminorex, la fenfluramine ou le benfluorex, cocaïne, amphétamines, imatinib, interféron), à des maladies (HIV, hypertension portale, schistostomiase, sclérodermie), à l’obésité ou à l’âge ; vu sa fréquence (200 millions d’humains), la schistostomiase est probablement la principale cause d’HTAP dans le monde.
- 1’ - Maladie pulmonaire veino-occlusive.
- 1’’ - HTAP persistante du nouveau-né (2 :1'000 bébés).
- 2 - Hypertension pulmonaire (HTP) postcapillaire (P télédiast VG > 18 mmHg, PAPO > 15 mmHg, mais RAP < 3 U Wood et gradient transpulmonaire < 12 mmHg) : défaillance systolique ou insuffisance diastolique restrictive du VG, valvulopathie mitrale ; c’est la cause la plus fréquente d’HTP chez l’adulte (65% des cas) [10,42].
- 3 - HTAP associée à l’hypoxie alvéolaire : BPCO, emphysème, SDRA, apnée du sommeil (SAS), hypoxie d’altitude, PEEP excessive ; l’élévation de la PAP est en général modérée (PAPm 25-35 mmHg).
- 4 - HTAP liée à la maladie thrombo-embolique pulmonaire chronique ; 4% des embolies pulmonaires aiguës se soldent par une non-résorption des thrombi.
- 5 - HTAP d’origine multifactorielle non éclaircie (sarcoïdose, histiocytose X, maladie de Gaucher, anémie hémolytique, maladies myéloprolifératives, insuffisance rénale dialysée).
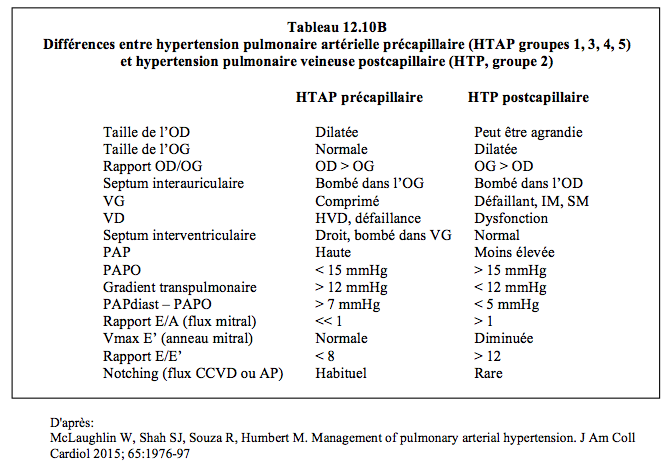
Dans les groupes 1, 3, 4 et 5, l’HTAP est précapillaire (Tableau 12.10B) ; la POG et la PAPO sont normales (PAPO < 15 mmHg), et le gradient transpulmonaire (GTP = PAPm – POG, ou PAPm - PAPO) est > 12-15 mmHg ; les RAP sont élevées (> 240 dynes•s•cm-5 ou > 3 U Wood) (voir Figure 5.123) [32].
L’HTAP chronique présente une progression dans les lésions anatomo-pathologiques, typiques d’une panvasculopathie (voir Figure 5.124) [33].
- Hyperplasie intimale, épaississement des cellules endothéliales ;
- Hypertrophie de la média et muscularisation réversible des portions terminales de l’arbre pulmonaire ;
- Prolifération des fibroblastes adventitiels ;
- Infiltration de cellules inflammatoires ;
- Occlusion progressive et thrombose des petits vaisseaux ;
- Lésions plexiformes d’angioprolifération dans l’HTAP idiopathique oblitérant les artérioles ; cette lésion est absente dans l’HTP postcapillaire.
L’HTP liée aux pathologies du cœur gauche (groupe 2) est la catégorie de loin la plus fréquente d’hypertension pulmonaire ; la moyenne d’âge y est plus élevée. Dans ce groupe, l’HTP est primairement postcapillaire : POG et PAPO élevées (> 15 mmHg), RAP et gradient transpulmonaire bas (GTP < 15 mmHg). Le gradient entre la PAPdiast et la PAPO est < 5 mmHg; il est plus sensible et plus spécifique que le GTP [10,13,42]. Cette élévation passive de la PAP est liée à l’augmentation de pression dans l’OG et dans les veines pulmonaires ; elle s'accompagne d'un exsudat intersticiel et d'un dysfonctionnement des pompes Na+-K+ qui extraient l'eau des alvéoles [14]. Elle est réversible si la lésion gauche est corrigée (chirurgie de la valve mitrale, correction de la défaillance du VG). Cependant, cette HTP, qui induit aussi une dysfonction endothéliale avec altération de la production de NO et d’endothéline, est souvent accompagnée d’une vasoconstriction artériolaire réactionnelle (HTAP) qui augmente la PAP hors de proportion avec l’élévation de la pression veineuse pulmonaire (gradient transpulmonaire > 12 mmHg dans 45% des cas de PAPO > 15 mmHg). La finalité de cette vasoconstriction due au remodelage artériolaire est probablement de diminuer l’engorgement pulmonaire en freinant le débit : l’administration de vasodilatateurs pulmonaires (NO, époprosténol, bosentan) dans cette situation tend à augmenter la congestion capillaire et la POG et n’améliore pas le débit cardiaque ; elle peut même conduire à l’OAP [42]. Seuls les anti-phospho-diestérases-5 comme le sildénafil semblent améliorer la symptomatologie et baisser la PAP [16]. La présence d’une HTAP précapillaire dans le cadre des maladies du cœur gauche en aggrave significativement le pronostic et en triple la mortalité [10,27]. Cette combinaison d'HTP pré- et post-capillaire est caractérisée par un gradient diastolique (PAPdiast – PAPO) > 7 mmHg, qui est lui-même est un prédicteur indépendant de mortalité [13]. Bien que la correction chirurgicale des lésions gauches puisse normaliser la pression veineuse pulmonaire, cette vasoconstriction artérielle persiste dans le postopératoire immédiat, mais elle tend à diminuer avec le temps. Lors de défaillance diastolique du VG (insuffisance gauche restrictive à fraction d’éjection préservée), l’HTP est présente dans 80% des cas. La prévalence de l’HTP est de 40% dans l’insuffisance gauche à FE abaissée. Elle est de 75% dans la sténose mitrale et d’environ 30% dans l’insuffisance mitrale [18]. La mortalité augmente de 30% par 10 mmHg d’élévation de la PAPsyst : à 3 ans, elle est de 50% si la PAPs est ≥ 50 mmHg, mais de 27% si la PAPs est < 48 mmHg [23]. La difficulté pour le VD d’assurer la perfusion pulmonaire en cas d’HTAP fait que des RAP > 5 U Wood et un gradient transplmonaire > 15 mmHg sont des contre-indications à la transplantation cardiaque [31].
Le pronostic de l’HTAP est très réservé. Sans traitement, la survie moyenne est de 2.8 ans, seuls 34% des patients atteignent 5 ans ; chez les congénitaux, la survie est 2-3 fois supérieure [1,40]. Sous traitement, la survie globale à 1, 3, 5 et 7 ans est respectivement de 88%, 70%, 60% et 55% [2]. Dans toutes les affections auxquelles elle est associée, l’hypertension pulmonaire est un facteur aggravant majeur. En chirurgie, elle augmente de 2 à 4 fois la mortalité opératoire [21]. D’une manière générale, la survie est la plus faible dans l’HTAP de la maladie thromboembolique (survie à 5 ans sans chirurgie : 55%). Elle est meilleure dans l’HTAP primaire (survie à 5 ans : 66%) et la plus élevée dans les cardiopathies congénitales (survie à 5 ans : > 90%) [21,35]. En effet, les deux ventricules ont une masse égale pendant la vie intra-utérine ; s’il est exposé dès la naissance à une postcharge élevée, le VD conserve une structure fœtale et s’hypertrophie parallèlement au VG [6]. La mortalité n’est pas associée à la valeur de la PAP en elle-même, mais à l’étiologie, à la défaillance du VD et à la valeur des RAP (> 10 unités Wood) ou de la compliance.
Physiopathologie
En systole, l’arbre vasculaire pulmonaire doit absorber la totalité du volume systolique car la valve mitrale est fermée, raison pour laquelle les RAP sont dix fois plus basses que les RAS. Lorsque le débit cardiaque augmente à l’effort, les résistances vasculaires pulmonaires baissent afin de contenir cet excès de volume, mais la diminution des RAP ne peut pas être importante vu que le lit pulmonaire est déjà en vasodilatation active au repos. La PAP s’élève donc à l’effort. Une augmentation excessive de la PAP à l’exercice est un signe précoce de l’HTAP. Par contre, son augmentation au repos est un signe tardif puisqu’il faut que 50% de la circulation soit obstruée pour qu’elle reste élevée en permanence [26]. La postcharge du VD consiste en trois composantes : la résistance artériolaire fixe, l’impédance pulsatile et la compliance vasculaire. En clinique, on ne calcule que la première (RAP = (PAPm – PAPO)/DC), qui mesure la résistance moyenne comme si le flux était continu ; elle représente 60-70% de la résistance à l’éjection [24,43]. La deuxième quantifie la postcharge dynamique du VD puisqu’elle traduit l’opposition au flux dans un système pulsatile (Z = Pinst/flux) ; elle peut se calculer à partir du flux Doppler pulsé dans l’AP et de la mesure simultanée de la PAP. Elle représente le tiers de la postcharge du VD. Comme elle inclut la résistance et la rigidité des vaisseaux pulmonaires, elle offre une meilleure corrélation avec la survie que les RAP seules [20]. La compliance est, de manière simplifiée, le rapport entre le volume systolique et la pression pulsée (C = VS/PP, où PP = PAPs – PAPd). Contrairement à la circulation systémique où elle est localisée dans l’aorte et les grandes artères élastiques, la compliance pulmonaire est le fait de tout l’arbre vasculaire réparti à travers les poumons. L’arbre pulmonaire est donc un système à basse résistance et à haute compliance. Lorsque cette dernière diminue, la différentielle systolo-diastolique, ou pression pulsée, augmente. Comme la compliance et la résistance varient en sens inverse, leur produit (R • C, en 1/seconde) reste constant ; il représente la constante de temps qui caractérise la baisse de la PAP en diastole [24] ; il reste stable au cours du traitement de l’HTAP, alors que l’évolution naturelle de la maladie entraîne une baisse de compliance plus importante que l’augmentation des RAP. La compliance est donc un marqueur plus fin du degré d’HTAP [25] : un rapport VS/PP < 0.81 mL/mmHg prédit une probabilité de survie à 4 ans de < 40%, alors qu’un rapport > 2.0 prédit une survie de 100% [28]. D’ailleurs, la rigidité de l’arbre vasculaire (baisse de la compliance) est le principal déterminant lié à la diminution des indices de fonction du VD [37]. La relation qui unit la résistance et la compliance n’est toutefois pas linéaire, mais curvilinéaire et logarithmique [25]. Ainsi de petites variations de résistance se traduisent par de larges variations de la compliance lorsque les RAP sont basses et que la courbe est redressée, alors que les mêmes variations de résistance modifient peu la compliance lorsque les RAP sont élevées parce que la courbe est assez plate. Les vasodilatateurs pulmonaires sont donc plus efficaces au début de la maladie qu’en phase terminale. La silhouette de la courbe de pression systolique en AP illustre le degré de rigidité des vaisseaux pulmonaires (Figure 12.28A) : on voit une augmentation de la pression pulsée et de la pression de réflexion (pression réfléchie : voir Chapitre 5 Couplage ventriculo-artériel) caractéristique de l’HTAP ; la deuxième s’ajoute à la pression d’éjection du VD et augmente donc sa postcharge réelle [7].

Figure 12.28A : Couplage entre le VD et la circulation pulmonaire. La réflexion de l’onde de pression due à la rigidité de l’arbre pulmonaire et à l’élévation des RAP donne lieu à une augmentation significative (ΔP) de la pression systolique par rapport à la pression avec laquelle le flux est éjecté du VD dans l’AP (Pe, pression d’entrée); cette augmentation de pression élève la postcharge du VD. L’index d’augmentation (ΔP/PP) s‘accroît avec la rigidité des vaisseaux. Le crochetage Pe correspond au “notching” typique de l’HTAP sur le flux Doppler dans l’AP (échocardiographie) [7].
Le couplage ventriculo-artériel droit peut s'exprimer en clinique par le rapport entre une mesure de la fonction éjectionnelle du VD et la pression pulmonaire systolique, par exemple par le rapport TAPSE/PAPsyst (TAPSE: tricuspid annular plane systolic excursion). Ce dernier permet une stratification du pronostic au sein de la constellation des différentes formes d'insuffisance cardiaque (voir Figure 5.35) [14].
Les vaisseaux pulmonaires, moins innervés que le circuit systémique, ont une répartition différente des récepteurs sympathiques ; les récepteurs α y sont rares et les récepteurs β prédominent. La stimulation sympathique a un effet préférentiellement β vasodilatateur lorsque les RAP sont basses, mais un effet α vasoconstricteur lorsque les RAP sont déjà élevées [38]. Les vasoconstricteurs comme la nor-adrénaline ou la phényléphrine ont moins d’effet sur la circulation pulmonaire que sur la circulation systémique [34]. La vasopressine n’augmente pas la PAP car le rapport entre les récepteurs V1 (vasoconstriction) et V2 (vasodilatation) est en faveur des seconds dans le lit pulmonaire; elle a un effet vasodilatateur pulmonaire à faible dosage, mais un effet vasoconstricteur seulement à forte dose [9]. Les inhibiteurs de la phosphodiestérase-3 (IPDE3) comme la milrinone agissent sur les artérioles précapillaires et préalvéolaires qui contiennent l'enzyme PDE-3; ils y induisent une vasodilatation. Cependant, cet effet peut être atténué dans les artérioles remaniées des patients souffranat d'HTAP [39]. Les vaisseaux pulmonaires sont maintenus dans une vasodilatation active permanente, qui est la résultante d’un équilibre dynamique entre plusieurs éléments :
- NO• : vasodilatateur sécrété localement par l’endothélium en fonction de la pulsatilité;
- Prostacycline I2E1 : vasodilatation ;
- Endothéline E1 : vasoconstriction ;
- Thromboxane A2, sérotonine et angiotensine II : vasoconstriction ;
- Hypoxie alvéolaire (PaO2 < 60 mmHg) : vasoconstriction locale ;
- Hypercapnie et acidose (élévation de la [H+] locale) : vasoconstriction.
Les vasodilatateurs pulmonaires dimiuent également l’adhésivité plaquettaire, alors que les vasoconstricteurs l’augmentent ; de plus, les vasoconstricteurs favorisent la prolifération cellulaire endothéliale, myoblastique et fibroblastique. Dans l’hypertension pulmonaire, la production de NO• est freinée, et l’activité des phosphodiestérases-5, qui catabolisent le cGMP, est augmentée. Or ce dernier est le messager intracelulaire du NO• ; il abaisse la [Ca2+]i dans les cellules musculaires lisses et provoque une vasodilatation. Après une CEC, la production pulmonaire de NO• et de prostacycline diminue à cause d’une série de phénomènes : syndrome inflammatoire, lésion d’ischémie/reperfusion sur la circulation bronchique, protamine, etc. La noradrénaline étant métabolisée par l’endothélium pulmonaire, ses concentrations sont plus élevées en cas d’hypertension pulmonaire [22].
La vasoconstriction pulmonaire hypoxique détourne le flux des zones mal ventilées vers celles qui le sont bien ; elle améliore l’appariement ventilation – perfusion et restreint l’effet shunt conduisant à la désaturation artérielle (voir Figure 5.126). Par ce phénomène, une hypoxémie persistante conduit à une hypertension pulmonaire chronique. L’HTAP chronique présente une progression dans les lésions anatomo-pathologiques : muscularisation réversible des portions terminales de l’arbre pulmonaire et hypertrophie de la média, prolifération des fibroblastes adventitiels, puis épaississement des cellules endothéliales et enfin lésions plexiformes et fibrose (voir Figure 5.124) [33]. Au stade terminal, l’HTAP sévère est fixée (PAPm > 55 mmHg, RAP > 10 U Wood ou > 800 dynes•s•cm-5) et n’est plus modulable. Dans les cardiopathies congénitales sur débit pulmonaire excessif (shunt G-D), la PAP voisine la PA systémique et le shunt devient bidirectionnel ; on parle de syndrome d’Eisenmenger [32]. L’arbre pulmonaire est alors constellé de thrombus muraux, d’où l’importance de l’anticoagulation.
Le ventricule droit
La circulation pulmonaire et le ventricule droit forment un tout fonctionnel qu’il est important d’étudier ensemble en fonction du couplage ventriculo-artériel [7]. La survie des patients souffrant d’HTAP dépend entièrement de la fonction du VD, car il faut la conjonction de deux éléments pour atteindre de hautes valeurs de PAP : une impédance artérielle et des résistances artériolaires élevées d’une part, et une force propulsive suffisante de l’autre. Celle-ci est fournie par le VD, dont la taille et l’hypertrophie ont une forte valeur pronostique pour la survie des patients. Ainsi, l’HTAP est fonction de la capacité du VD à générer chroniquement des pressions pulmonaires élevées. Une PAP de 90/50 mmHg signifie que le VD est capable de soutenir ce régime de pression. La défaillance droite se traduit au contraire par l’impossibilité de travailler contre une telle postcharge : la PAP tend à redescendre, alors que la situation hémodynamique empire (Figure 12.28B) [16]. La gravité et le pronostic de la maladie tiennent donc davantage à la fonction ventriculaire droite qu’à la valeur de la PAP en elle-même.
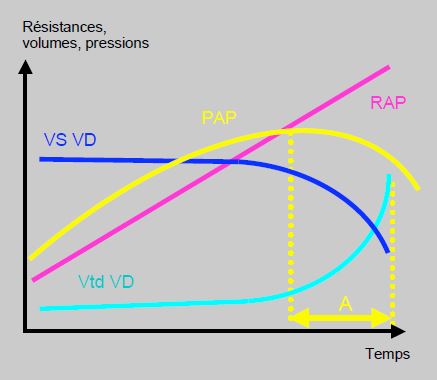
Figure 12.28B : Couplage entre le VD et la circulation pulmonaire. Schématisation de la relation entre les résistances artérielles pulmonaires (RAP), le volume systolique du VD (VS VD), le volume télédiastolique du VD (VtdVD) et la pression artérielle pulmonaire (PAP). L’HTAP est fonction de la capacité du VD à générer chroniquement des pressions pulmonaires élevées. Dès que le VD défaille, la PAP tend à redescendre, alors que la situation hémodynamique empire et que les RAS continuent à augmenter. Dans le laps de temps A (en jaune), la PAP mesurée est plus basse que précédemment, non par amélioration de la situation mais pas péjoration de fonction du VD. La gravité clinique et le pronostic de la maladie tiennent donc davantage à la fonction ventriculaire droite qu’à la valeur de la PAP en elle-même [16].
En situation aiguë, un VD normal peut soutenir une PAPsyst de 40-60 mmHg pendant 1-2 heures avant de défaillir et de se dilater. En situation chronique et progressive, l’adaptation du VD à une augmentation de sa postcharge se fait par une hypertrophie concentrique et une transformation vers une forme plus arrondie qui le rendent davantage dépendant de la précharge et moins de la postcharge : plus il s’hypertrophie, plus le VD ressemble au VG (voir Chapitre 5 Dysfonction du VD). Cette adaptation lui permet d'augmenter sa contractilité de 4 à 5 fois [44]. Mais les limites de ce processus sont vite atteintes, car le VD ne peut surmonter longtemps un excès de résistance à l’éjection et il commence à se dilater pour utiliser au aximum l'effet Starling, ce qui augmente encore son stress de paroi et sa consommation d’O2, alors que sa perfusion coronarienne est davantage compromise [4]. La dilatation l’amène en butée contre le péricarde. Comme l’espace intrapéricardique est limité, l’augmentation de volume du VD fait basculer le septum interventriculaire dans la cavité gauche en diastole et limite le remplissage diastolique de celle-ci. Ainsi une surcharge de volume aggrave non seulement l’insuffisance congestive droite mais encore limite le remplissage et le volume d’éjection du VG. L’interaction diastolique par le déplacement du septum est au moins aussi importante que la chute de l'éjection du VD dans la genèse du bas débit systémique [6]. Avec l’hypertrophie et l’élévation de postcharge, la durée de la contraction droite se prolonge et le VD se contracte encore lorsque le VG est déjà en diastole, ce qui produit une bascule systolique du septum vers la gauche et une dyskinésie septale qui gène l’éjection des deux ventricules [43]. L’augmentation de la postcharge gauche par un vasoconstricteur tend au contraire à repousser le septum dans sa position normale convexe dans le VD, donc à retrouver l’appui du VG dans l’éjection droite. La dilatation du VD (> 85 mL/m2, VS/VtsVD > 1) et de l’OD (S > 15 cm2/m, où m = hauteur de l’individu en mètre), l’élévation de la PVC (> 15 mmHg) et la baisse de la fonction systolique droite (FE < 0.35, voir Insuffisance ventriculaire droite) sont les principaux critères autorisant une évaluation pronostique [44]. L’insuffisance cardiaque droite se caractérise par un bas débit et par une élévation de la pression veineuse centrale au repos, avec rétention liquidienne, stase veineuse, oedèmes, ascite, hépatopathie et insuffisance rénale. L’élévation de la POD > 15 mmHg peut rouvrir un foramen ovale perméable et occasionner un shunt droite – gauche cyanogène. La mortalité de la défaillance droite dans le cadre de l’HTAP s’élève jusqu’à 40% [17]. De plus, certains vasodilatateurs pulmonaires comme les anti-endothéline-1 (bosentan) ont un effet inotrope négatif sur le VD [4].
Hémodynamique
Les patients souffrant d’hypertension pulmonaire se caractérisent par une perte complète de la compliance hémodynamique dans la circulation droite. Ils présentent une physiopathologie particulière [3,8,11].
- Le débit pulmonaire est abaissé et relativement fixe ; il ne peut pas augmenter proportionnellement à la demande en O2, d’où cyanose à l’effort. Toute élévation du débit cardiaque se traduit par une élévation importante de la PAP, pour autant que le VD ne soit pas dysfonctionnel.
- Face à l’augmentation chronique de sa postcharge, le VD dilate et s’hypertrophie (HVD) (Vidéo). Plus il est hypertrophié, plus le VD se comporte comme le VG ; il tolère l’augmentation de postcharge mais son débit devient dépendant de la précharge ; il ne peut plus amortir les variations du retour veineux en maintenant le débit pulmonaire constant ; l’hypovolémie conduit à une baisse du débit pulmonaire et à une hypoxémie.
Vidéo: Hypertrophie ventriculaire droite concentrique sur excès chronique de postcharge.
- En diastole, la pression du VD hypertrophié et surchargé est supérieure à celle du VG ; le septum interventriculaire bombe dans le VG et réduit le remplissage diastolique gauche (Figure 12.8A) (Vidéo) ; l’élévation de la postcharge gauche (vasoconstriction artérielle systémique) tend à replacer le septum dans sa position physiologique.
Vidéo: Défaillance et dilatation du VD avec dilatation massive de l'OD dans le cadre d'embolie pulmonaire; les cavités gauches sont comprimées par le basculement du septum interventriculaire et du septum interauriculaire.
- Une hypotension systémique peut compromettre la perfusion coronarienne droite en réduisant la composante systolique du flux coronaire vers le VD ; un vasoconstricteur systémique est requis pour parer au risque ischémique.
- Si le foramen ovale est perméable, un shunt droite ⟶ gauche cyanogène peut s’installer à la faveur d’une augmentation excessive de la POD (Vidéo).
Vidéo: Passage droit - gauche par un foramen ovale perméable; la POD est supérieure à la POG comme le démontre le bombement du septum interauriculaire; le flux couleur s'étend largement dans l'OG.
- Malgré l’épaississement des parois artérielles pulmonaires, les petits vaisseaux artériolaires périphériques conservent une réactivité vasculaire ; les RAP peuvent encore augmenter par hypoxémie, hypercarbie, acidose, hypothermie ou stress sympathique.
Lors de la prise en charge de ces malades en salle d'opération ou aux soins intensifs, il est capital d'éviter toutes les situations qui peuvent aggraver les RAP:
- Hypoventilation (hypercarbie, hypoxémie, atélectasies) ;
- Surpression intrathoracique (variable selon la fonction du VD, respecter une hyperventilation normobarique) ;
- Acidose ;
- Hypothermie ;
- Stimulation sympathique (stress, douleur, anxiété) ;
- Anémie aiguë (seuil de transfusion Hb > 90 gm/L) ; la baisse du transport d’O2 ne peut être compensée que par une augmentation du débit pulmonaire, qui élève considérablement la PAP.
| Hypertension pulmonaire (HTP) |
|
Définition de l'hypertension artérielle pulmonaire (HTAP): PAP moy > 25 mmHg au repos et RAP > 240 dynes•s•cm-5 (valeur normale : 60-120 dynes•s•cm-5, < 2 U Wood).
Classification de l'OMS:
- HTAP essentielle: HTAP primaire, HTAP des cardiopathies congénitales, HTAP secondaire (médicaments, obésité, âge)
- HTP postcapillaire (insuffisance ventriculaire gauche, valvulopathie mitrale)
- HTAP due à l'hypoxie alvéolaire (BPCO, SAS, haute altitude)
- Maladie thrombo-embolique pulmonaire
- HTAP d’origine multifactorielle non éclaircie
Syndrome d'Eisenmenger: HTAP fixée aréactive, PAPm > 55 mmHg, RAP > 800 dynes•s•cm-5.
Impact clinique de l'HTP:
- Débit pulmonaire fixe, hypoxémie à l'effort ou lors d’hypovolémie
- Maintien de la réactivité des petits vaisseaux (vasoconstriction pulmonaire hypoxique)
- Hypertrophie du VD: débit droit dépendant de la précharge, intolérance à l'hypovolémie
- Risque ischémique du VD élevé en cas d'hypotension systémique
- Insuffisance diastolique du VG (effet Bernheim)
- Dilatation et décompensation du VD
- Shunt D → G si foramen ovale perméable
Le pronostic de l’HTP tient davantage à l’état fonctionnel du VD qu’à la valeur de la PAP.
|
© BETTEX D, CHASSOT PG, Janvier 2008, dernière mise à jour, Novembre 2019
Références
- BADESCH DB, CHAMPION HC, GOMEZ SANCHEZ MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2009; 54:S55-66
- BENZA RL, MILLER DP, BARST RJ, et al. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from REVEAL. Chest 2012; 142:448-56
- BLAISE G, LANGLEBEN D, HUBERT B. Pulmonary arterial hypertension. Anesthesiology 2003; 99:1415-32
- BOGAARD HJ, ABE K, VONK NOORDEGRAAF A, et al. The right ventricle under pressure. Cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009; 135:794-804
- BRAUN EB, PALIN CA, HOGUE CW. Vasopressin during spinal anesthesia in a patient with primary pulmnary hypertension treated with intravenous epoprostenol. Anesth Analg 2004; 99:36-7
- BRONICKI RA, BADEN HP. Pathophysiology of right ventricular failure in pulmonary hypertension. Pediatr Crit Care Med 2010; 11(Suppl):S15-S22
- CHAMPION HC, MICHELAKIS ED, HASSOUN PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit. Circulation 2009; 120:992-1007
- CHASSOT PG, BETTEX DA. Anesthesia and adult congenital heart disease. J Cardiothorac Vasc Anesth 2006; 20: 414-37
- CURRIGAN DA, HUGHES RJ, WRIGHT CE, et al. Vasoconstrictor responses to vasopressor agents in human pulmonary and radial arteries: an in vitro study. Anesthesiology 2014; 121:930-6
- FANG JC, DE MARCO T, GIVERTZ MM, et al. World Health Organization Pulmonary Hypertension Group 2: Pulmonary hypertension due to left heart disease in the adult – a summary statement from the Pulmonary Hypertension Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2012; 31:913-33
- FISCHER LG, VAN AH, BURKLE H. Management of pulmonary hypertension: physiological and pharmacological considerations for anesthesiologists. Anesth Analg 2003; 96:1603-16
- GALIÉ N, HOEPER MM, HUMBERT M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009; 30:2493-537
- GERGES C, GERGES M, LANG MB, et al. Diastolic pulmonary vascular pressure gradient: a predictor of prognosis in "out-of-proportion" pulmonary hypertension. Chest 2013; 143:758-66
- GUAZZI M, NAEIJE R. Pulmonary hypertension in heart failure. Pathophysiology, pathobiology, and emerging clinical perspectives. J Am Coll Cardiol 2017; 69:1718-34
- HADDAD F, COUTURE P, TOUSIGNANT C, DENAULT AY. The right ventricle in cardiac surgery, a perioperative perspective: I. Anatomy, physiology and assessment. Anesth Analg 2009; 108:407-21
- HADDAD F, DOYLE R, MURPHY DJ. HUNT SA. Right ventricular function in cardiovascular disease, Part II. Pathophysiology, clinical importance and management of right ventricular failure. Circulation 2008; 117:1717-31
- HADDAD F, FUH E, PETERSON T, et al. Characteristics and outcome after hospitalization for acute right heart failure in patients with pulmonary arterial hypertension. Circ Heart Fail 2011; 4:692-9
- HADDAD F, KUDELKO K, MERCIER O, et al. Pulmonary hypertension associated with left heart disease: Characteristics, emerging concepts, and treatment strategies. Progr Cardiovasc Dis 2011; 54:154-67
- HOEPER MM, BOGAARD HJ, CONDLIFFE R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013; 62:D42-50
- HUNTER KS, LEE PF, LANNING CJ, et al. Pulmonary vascular input impedance is a combined measure of pulmonary vascular resistance and stiffness and predicts clinical outcomes better than pulmonary vascular resistance alone in pediatric patients with pulmonary hypertension. Am Heart J 2008; 155:166-74
- KIM NH, DELCROIX M, JENKINS DP, et al. Chronic thromboembolic pulmonary hypertension. J Am Coll Cardiol 2013; 62:D92-9
- KWAK YL, LEE CS, PARK YH, et al. The effect of phenylephrine and norepinephrine in patients with chronic pulmonary hypertension. Anaesthesia 2002; 57:9-14
- LAM CSP, ROGER VL, RODEHEFFER RJ, et al. Pulmonary hypertension in heart failure with preserved ejection fraction. A community-based study. J Am Coll Cardiol 2009; 53:1119-26
- LANKHAAR JW, WESTERHOF N, FAES TJC, et al. Quantification of right ventricular afterload in patients with and without pulmonary hypertension. Am J Physiol Heart Circ Physiol 2006; 291:H1731-H1737
- LANKHAAR JW, WESTERHOF N, FAES TJC, et al. Pulmonary vascular resistance and compliance stay inversely related during treatment of pulmonary hypertension. Eur Heart J 2008; 29:1688-95
- LAU EMT, MANES A, CELERMAJER DS, GALIE N. Early detection of pulmonary vascular disease in pulmonary arterial hypertension: time to move forward. Eur Heart J 2011; 32:2489-98
- LIM GB. Prognostic relevance of pulmonary hypertension in valvular disease. Nat Rev Cardiol 2015; 12:194
- MAHAPATRA S, NISHIMURA RA, SORAJJA P, et al. Relationship of pulmonary arterial capacitance and mortality in idiopathic pulmonary arterial hypertension. J Am Coll Cardiol 2006; 47:799-803
- MARON BA, HESS E, MADDOX TM, et al. Association of borderline pulmonary hypertension with mortality and hospitalization in a large patient cohort: insights from the Veteran Affairs Clinical Assessment Reporting and Tracking Program. Circulation 2016; 133:1240-8
- McLAUGHLIN W, ARCHER SL, BADESCH DB et al. ACC/AHA 2009 expert consensus document on pulmonary hypertension: A report of the American College of Cardiology Foundation Task Force on expert consensus documents and the American Heart Association. Circulation 2009; 119:2250-94
- MEHRA MR, KOBASHIGAWA J, STARLING R, et al. Listing criteria for heart transplantation: International Society for Heart and Lung Transplantation guidelines for the care of cardiac transplant candidates – 2006. J Heart Lung Transplant 2006; 25:1024-42
- OPOTOWSKY AR. Clinical evaluation and management of pulmonary hypertension in the adult with congenital heart disease. Circulation 2015; 131:200-10
- PALEVSKY HI, SCHLOO BL, PIETRA CC, et al. Primary pulmonary hypertension: Vascular structure, morphometry, and responsiveness to vasodilator agents. Circulation 1989; 80:1207-21
- RICH S, McLAUGHLIN VV. Pulmonary hypertension. In: ZIPES DP, et al, eds. Braunwald’s heart disease. A textbook of cardiovascular medicine. Philadelphia: Elsevier Saunders, 2005, 1807-42
- RUOHONIEMI DM, SISTA AK, DOANY CF, HEERDT PM. Perioperative pulmonary thromboembolism: current concepts and treatment options. Curr Opin Anaesthesiol 2018; 31:75-82
- SIMONEAU G, GATZOULIS MA, ADAIA I,, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62:D34-41
- STEVENS GR, GARCIA-ALVAREZ A, SAHNI S, et al. RV dysfunction in pulmonary hypertension is independently related to pulmonary artery stiffness. JACC Cardiovasc Inaging 2012; 5: 378-87
- SUBRAMANIAM K, YARED JP. Management of pulmonary hypertension in the operating room. Semin Cardiothor Vasc Anesth 2007; 11:119-36
- TABUCHI A, STYP-REKOWSKA B, SLUTSKY AS, et al. Precapillary oxygenation contributes relevantly to gas exchange in the intact lung. Am J Respir Crit Care Med 2013; 188:478-81
- THENAPPAN T, SHAH SJ, RICH S, et al. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J 2010; 35:1079-87
- TÜLLER C, NICOD LP. Hypertension pulmonaire chronique. Forum Méd Suisse 2008; 8:812-7
- VACHIÉRY JL, ADIR Y, BARBERÀ JA, et al. Pulmonary hypertension due to left heart disease. J Am Coll Cardiol 2013; 62:D100-8
- VONK-NOORDEGRAAF A, HADDAD F, CHIN KM, et al. Right heart adaptation to pulmonary arterial hypertension. J Am Coll Cardiol 2013; 62:D22-33
- VONK NOORDEGRAAF A, WESTERHOF BE, WESTERHOF N. The relationship between the right ventricle and its load in pulmonary hypertension. J Am Coll Cardiol 2017; 69:236-43
12 Anesthésie et insuffisance ventriculaire
- 12.1 Introduction
- 12.2 Présentation clinique
- 12.3 Traitement de l'insuffisance ventriculaire
- 12.4 Assistance ventriculaire
- 12.4.1 Principes, indications et physiopathologie
- 12.4.2 Contre-pulsion par ballon intra-aortique (CPIA)
- 12.4.3 Extra-Corporeal Membrane Oxygenation (ECMO)
- 12.4.4 Dispositifs d’assistance ventriculaire à court terme
- 12.4.5 Dispositifs à long terme
- 12.4.6 Problèmes liés à une assistance ventriculaire
- 12.4.7 Rôle de l’échocardiographie dans l’assistance ventriculaire
- 12.4.8 Anesthésie et assistance ventriculaire
- 12.5 Hypertension pulmonaire
- 12.6 Anesthésie en cas d'insuffisance ventriculaire
- 12.6.1 Contraintes
- 12.6.2 Evaluation préopératoire
- 12.6.3 Stratégies d’anesthésie
- 12.6.4 Ventilation et insuffisance ventriculaire
- 12.6.5 Agents intraveineux et halogénés
- 12.6.6 Anesthésie générale du patient en insuffisance ventriculaire gauche
- 12.6.7 Anesthésie loco-régionale
- 12.6.8 Insuffisance droite et hypertension pulmonaire
- 12.6.9 La thrombendartérectomie pulmonaire (TEAP)
- 12.7 Conclusion